
Feokromositoma, epinefrin, norepinefrin ve daha az oranda dopamin de dahil olmak üzere katekolaminlerin üretiminden sorumlu olan adrenal medullanın kromaffin hücrelerinden kaynaklanan nadir bir tümördür. Bu tümörler bu hormonları aşırı miktarda salgılayarak çeşitli klinik semptomlara yol açabilir. Feokromositomalar iyi huylu veya kötü huylu tümörler olarak ortaya çıkabilir ve vakaların yaklaşık %10’u kötü huyludur. Kötü huylu feokromositomalar vücudun diğer bölgelerine metastaz yapma potansiyeline sahiptir.
Klinik Özellikler:
Feokromositomalar tarafından aşırı katekolamin üretimi, aşağıdakiler de dahil olmak üzere önemli kardiyovasküler semptomlara yol açabilir:
- Hipertansiyon: Genellikle paroksismaldir, ancak sürekli de olabilir.
- Palpitasyonlar
- Taşikardi
- Baş ağrısı
- Terleme
- Anksiyete ve Panik Ataklar
- Flushing
Diğer semptomlar arasında kilo kaybı, hiperglisemi ve ortostatik hipotansiyon yer alabilir. Bazı vakalarda katekolamin kaynaklı kardiyomiyopati veya aritmiler gelişebilir.
Katekolaminler ve Dopamin Salgılanması:
- Katekolaminler:** Feokromositomalar öncelikle norepinefrin ve epinefrin salgılar, ancak kötü huylu vakalarda tümör, malignite için bir biyobelirteç görevi görebilecek önemli miktarda dopamin de salgılayabilir.
- Malignite:** Feokromositomaların çoğunluğu iyi huylu olsa da, yaklaşık %10’u kötü huyludur ve kemikler, karaciğer ve lenf düğümleri dahil olmak üzere uzak organlara yayılma potansiyeline sahiptir.
Teşhis:
Feokromositoma tanısı biyokimyasal testlere ve görüntüleme yöntemlerine dayanır. Temel tanısal testler şunları içerir:
24 Saatlik İdrar Katekolamin ve Metanefrin Testi:
- Katekolaminlerin (norepinefrin, epinefrin ve dopamin) ve metabolitlerinin (metanefrin ve normetanefrin) 24 saatlik idrar koleksiyonunda ölçülmesi, aşırı hormon salgılanmasını tespit etmek için hassas bir yöntemdir.
- İdrar Metanefrinleri:** Bunlar katekolaminlerin parçalanma ürünleridir ve katekolaminlerin kendilerinden daha stabil kabul edilirler, bu da onları tanı için tercih edilen bir belirteç haline getirir. Yüksek seviyeler feokromositomayı kuvvetle düşündürür.
Plazma Serbest Metanefrinleri:
- Bu, özellikle epizodik semptomları olan hastalarda alternatif bir testtir, çünkü metanefrinler hipertansif bir olaydan sonra plazmada katekolaminlerden daha uzun süre yüksek kalır.
Görüntüleme Çalışmaları:
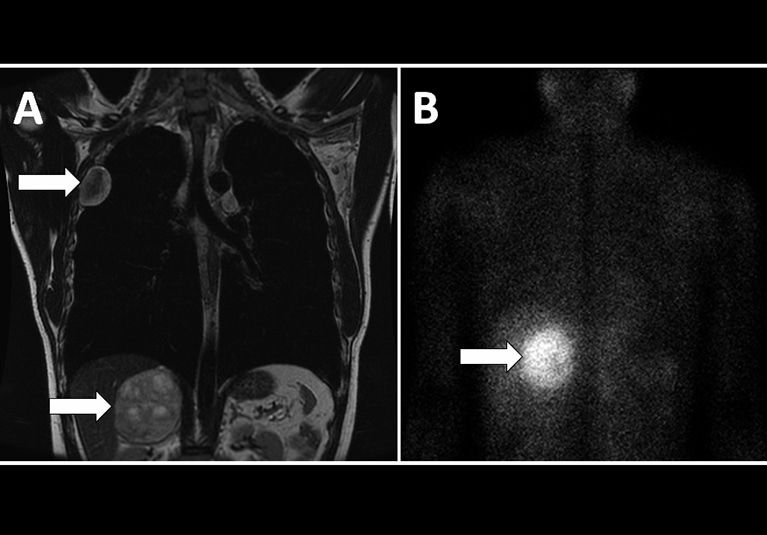
- BT (Bilgisayarlı Tomografi) veya MRI (Manyetik Rezonans Görüntüleme):** Bu görüntüleme teknikleri tümörü lokalize etmek için kullanılır. Feokromositomaların çoğu adrenal bezlerin içinde yer alır, ancak paraganglioma adı verilen adrenal dışı feokromositomalar sempatik zincir boyunca ortaya çıkabilir.
- MIBG Sintigrafisi (İyot-123-metaiodobenzilguanidin):** Bu nükleer tıp taraması metastatik veya ekstra adrenal feokromositomaları tespit etmek için yararlıdır.
- PET (Pozitron Emisyon Tomografisi): Malignite veya metastaz durumlarında, özellikle 18F-FDG kullanan PET taramaları, tümör yayılımını evrelemek ve değerlendirmek için kullanılabilir.
Genetik İlişkiler:
Feokromositomalar sporadik olarak veya kalıtsal sendromların bir parçası olarak ortaya çıkabilir. Feokromositomalar ile ilişkili genetik mutasyonlar şunları içerir:
- Multipl Endokrin Neoplazi Tip 2 (MEN2):** RET proto-onkogenindeki mutasyonlarla ilişkilidir.
- Von Hippel-Lindau (VHL) Hastalığı
- Nörofibromatozis Tip 1 (NF1)
- Süksinat Dehidrogenaz (SDHx) Mutasyonları: Paragangliomalar ve ailesel feokromositoma sendromları ile ilişkilidir.
Yönetim:
Feokromositoma için birincil tedavi tümörün cerrahi rezeksiyonudur, ancak katekolamin salınımının neden olduğu intraoperatif hipertansif krizleri önlemek için preoperatif tıbbi yönetim çok önemlidir. Ameliyat öncesi yönetim tipik olarak şunları içerir:
- Alfa-blokaj (Fenoksibenzamin veya Doksazosin):** Kan basıncını kontrol etmek ve hipertansif krizleri önlemek için.
- Beta-blokaj (Propranolol veya Atenolol): Kalp hızını ve aritmileri kontrol etmek için alfa-blokajdan sonra eklenir.
Malign veya metastatik feokromositomalar için ek tedaviler şunları içerebilir:
- Metastatik lezyonlar için Radyoterapi:
- Kemoterapi veya Hedefe Yönelik Tedavi: Bazen inoperabl veya metastatik hastalık için kullanılır.
- 131I-MIBG Tedavisi: Radyoaktif bileşiği emen metastatik feokromositomların tedavisinde kullanılır.

Keşif
Adrenal medullanın kromaffin hücrelerinden kaynaklanan nadir bir katekolamin salgılayan tümör olan feokromositoma, endokrinoloji ve cerrahideki daha geniş eğilimleri yansıtan zengin bir keşif ve tıbbi ilerleme geçmişine sahiptir.
İçindekiler
1. Katekolamin Kaynaklı Semptomların Erken Tanınması (19. Yüzyıl)
Feokromositomanın hikayesi 19. yüzyılda, tümörün kendisi net bir şekilde tanımlanmadan çok önce başlar. O dönemde doktorlar gizemli bir dizi semptom fark etmeye başladılar; kan basıncında şiddetli dalgalanmalar, şiddetli baş ağrıları, çarpıntı ve yoğun anksiyete. Bunlar bazen o zamanlar “paroksismal hipertansiyon” olarak adlandırılan duruma bağlanıyordu. Ancak gerçek neden anlaşılamamıştı. İlginç bir şekilde, bilim insanları adrenal fizyolojiyi anlamaya başlayana kadar bu semptomları katekolaminlerin aşırı üretimine bağlayamadılar.
1855 yılında Fransız fizyolog Charles-Édouard Brown-Séquard, böbreküstü bezlerinin sadece körelmiş organlar olmadığını, vücudun düzenlenmesinde kritik bir role sahip olduğunu tespit ederek bir çığır açtı. Bu keşfi feokromositoma ile ilişkilendirmemiş olsa da, çalışmaları daha sonraki gelişmelerin temelini atmıştır.
2. ‘Feokromositoma’ Teriminin Bulunuşu (1912)
“Feokromositoma” terimi Yunanca phaios (karanlık) ve chroma (renk) kelimelerinden türemiştir ve tümörün kromaffin hücrelerinin belirli boyalar altında koyu boyanmasına atıfta bulunmaktadır. Tümör ilk olarak 1912 yılında Alman patolog Ludwig Pick tarafından tanımlanmış ve Pick, tümör hücrelerinin krom tuzlarını absorbe etme yetenekleri nedeniyle koyu boyanma özelliklerine dikkat çekerek bu terimi kullanmıştır.
Bu dönem patoloji alanındaki keşifler açısından oldukça zengindi ve Pick’in çalışmaları, bilim insanlarının tümör biyolojisinin sırlarını ortaya çıkarmak için boyama tekniklerinden nasıl yararlanmaya başladığını gösteriyordu. Epinefrin ve norepinefrin salgılayan kromaffin hücreleri üzerine yaptığı gözlemler, feokromositomanın biyokimyasal doğasına dair ilk ipuçlarını verdi.
3. Katekolamin Salgılanması ve Feokromositoma Arasındaki Bağlantı (1926)
1920’lerde, katekolamin fizyolojisinin anlaşılmasındaki atılımlar feokromositoma teşhisi için kritik öneme sahipti. 1926 yılında İsviçreli patolog César Roux ve meslektaşı Charles Guillemin feokromositomaların katekolamin salgıladığını vurgulamış ve bu aşırı hormon seviyelerinin karakteristik semptomlara neden olduğunu açıklamıştır.
Bu, aşırı hormon salgılayarak tahribata yol açabilen bir “endokrin tümör” fikrinin klinik olarak ilk kez anlamlı hale geldiği zamandı. Roux ve Guillemin’in katkıları feokromositomanın gizemli ve ölümcül bir durumdan teşhis edilebilir ve nihayetinde tedavi edilebilir bir hastalığa dönüşmesine yardımcı oldu.
4. İlk Cerrahi Çıkarma (1926)
1926 yılında, feokromositomanın ilk cerrahi olarak çıkarılması Mayo Klinik’te Charles Mayo tarafından gerçekleştirilmiştir. Çığır açan bu ameliyat, semptomların ciddiyetine rağmen tümörün rezeksiyon yoluyla tedavi edilebileceğini gösterdi. Mayo’nun ameliyat notlarında yer alan anekdotlar bu ameliyatların dramatik doğasını vurgulamaktadır – yıllarca paroksismal hipertansiyondan muzdarip olan hastalar aniden semptomlarından kurtulmuşlardır.
Kayda değer vakalardan biri, ani şiddetli hipertansiyon, taşikardi ve anksiyete nöbetleri geçiren genç bir kadındı. Mayo adrenal tümörünü başarılı bir şekilde çıkardıktan sonra, kan basıncı normale döndü ve normal bir hayat yaşamaya başladı. Bu başarı öyküsü, feokromositoma tedavisinde cerrahinin öneminin altını çizdi ve daha cesur prosedürlere kapı açtı.
5. Biyokimyasal Teşhisin Gelişimi (1950’ler)
20. yüzyılın ortalarında araştırmacılar feokromositomanın tanısal doğruluğunu geliştirmeye odaklanmaya başladı. Büyük bir atılım, idrarda katekolamin testinin geliştirilmesiyle gerçekleşti. 1950’lerde araştırmacılar katekolamin metabolitlerinin, özellikle de metanefrinlerin idrarda ölçülebileceğini ve güvenilir bir teşhis belirteci olarak kullanılabileceğini keşfetti.
O döneme ait anekdotlar, kliniklere esrarengiz semptomlarla gelen hastaları ve doktorların idrarda katekolaminleri ölçme ve tanıyı doğrulama konusunda yeni keşfedilen yeteneğe nasıl hayret ettiklerini anlatıyor. Bu, klinisyenlerin şüpheli feokromositoma vakalarına yaklaşımında devrim yaratmış ve keşif ameliyatlarına olan ihtiyacı azaltmıştır.
6. Malign Feokromositoma ve Dopamin (1970’ler)
Malign feokromositoma anlayışı 20. yüzyılın ikinci yarısı boyunca gelişmiştir. Feokromositomaların çoğu iyi huylu olsa da yaklaşık %10’unun kötü huylu olduğu anlaşıldı. Dahası, bu kötü huylu tümörler genellikle iyi huylu feokromositomalar tarafından tipik olarak yüksek miktarlarda salgılanmayan bir hormon olan dopamin üretiyordu.
1970’lerde, kötü huylu vakaları inceleyen araştırmacılar, hasta idrar örneklerinde daha yüksek seviyelerde dopamin metabolitleri fark etmeye başladı. Eisenhofer ve arkadaşları tarafından 20. yüzyılın sonlarında yapılan dönüm noktası niteliğindeki bir çalışma, yüksek dopamin seviyelerinin feokromositomalarda malign davranışla güçlü bir şekilde ilişkili olduğunu vurgulayarak daha doğru prognoz ve tedavi planlamasına olanak sağladı.
7. Genetik Anlayış ve Sendromik Bağlantılar (20. Yüzyılın Sonları)
Feokromositoma ile ilişkili genetik mutasyonların keşfi bir başka önemli dönüm noktası olmuştur. 20. yüzyılın sonlarında, feokromositomaların Multipl Endokrin Neoplazi tip 2 (MEN2), von Hippel-Lindau (VHL) hastalığı ve Nörofibromatozis tip 1 (NF1) gibi kalıtsal sendromların bir parçası olabileceği anlaşılmıştır.
MEN2 sendromunda RET proto-onkogen mutasyonunun keşfi en kayda değer gelişmelerden biriydi. Bu, feokromositomanın genetik temeline ilişkin değerli bilgiler sağlamış ve risk altındaki bireylerin genetik olarak test edilmesine ve erken taranmasına olanak tanımıştır. Bu sendromlardan etkilenen aileler artık feokromositomaları erken tespit etmek için bir araca sahipti ve doktorların kalıtsal risk yönetimine yaklaşımını değiştirdi.
8. Modern Görüntüleme ve Tedavi (21. Yüzyıl)
- yüzyılda PET-BT taramaları ve MIBG sintigrafisi gibi sofistike görüntüleme tekniklerinin ortaya çıkması, klinisyenlerin feokromositomların yerini doğru bir şekilde tespit etmesine ve malign vakalarda metastazı değerlendirmesine olanak sağlamıştır. Bu görüntüleme yöntemlerinin geliştirilmesi, hem tanı hem de cerrahi planlama için oyunun kurallarını değiştiren bir gelişme olarak tanımlanmıştır.
Klinik uygulamadan yeni bir anekdot, dirençli hipertansiyonu olan orta yaşlı bir adamla ilgiliydi. Kan testleri normal olmasına rağmen, semptomları güçlü bir şekilde feokromositoma olduğunu düşündürüyordu. Bir PET-BT taraması, adrenal bezinde küçük bir tümör tespit etti ve daha sonra laparoskopik cerrahi ile başarıyla çıkarıldı ve modern görüntülemenin sağladığı hassasiyeti gösterdi.
İleri Okuma
- Eisenhofer, G., Lenders, J. W. M., Timmers, H., et al. (2004). “Biochemical Diagnosis of Pheochromocytoma: How to Distinguish True- from False-Positive Test Results.” Journal of Clinical Endocrinology & Metabolism, 88(6), 2656–2666. doi:10.1210/jc.2004-0192.
- Lenders, J. W. M., Eisenhofer, G., Mannelli, M., & Pacak, K. (2005). “Pheochromocytoma.” The Lancet, 366(9486), 665–675. doi:10.1016/S0140-6736(05)67139-5.
- Young, W. F. (2007). “Pheochromocytoma and Paraganglioma.” Journal of Clinical Endocrinology & Metabolism, 92(2), 356–365. doi:10.1210/jc.2006-1690.
- Amar, L., & Plouin, P. F. (2013). “Recent Advances in the Genetics of Pheochromocytoma.” Annales d’Endocrinologie, 74(2), 97–106. doi:10.1016/j.ando.2013.03.004.
- Fishbein, L., Leshchiner, I., Walter, V., et al. (2017). “Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma.” Cancer Cell, 31(2), 181–193. doi:10.1016/j.ccell.2017.01.001.