- Kalıtsal olarak belirli proteinlerin sentezlenememesi sebebiyle kan pıhtılaşması engellenir. Kişi kanamaya daha yatkın hale gelir. Kelimenin kökeni de bu yatkınlığı ifade eder. (Bkz; Hem-o-fili)
- X krozomal resesif bir hastalıktır.
Hemofili, Von Wille brand sendromundan sonra en sık görülen kalıtsal kan pıhtılaşma bozukluğu olmasına rağmen nadir görülen bir hastalıktır. Bununla birlikte, bu hastalığın Avrupa’nın kraliyet ailelerinde görülme sıklığının artması, son yüzyıllarda kötü şöhretine katkıda bulunmuştur, bu nedenle hastalık kraliyet hastalığı olarak bilinir hale gelmiştir. İngiltere Kraliçesi Victoria kraliyet hastalığını oğlu Prens Leopold’a geçirdi. Prens Leopold, daha sonra başından yaralanarak öldü. Kraliçe’nin kızları aracılığıyla hemofili İspanyol, Prusya ve Rus hanedanlarına da yayıldı.
Hemofili, pıhtılaşma faktörleri FV ve FV2’nin X’e bağlı resesif kalıtımsal bir eksikliğini tanımlar. Pıhtılaşma faktörleri FVIII (hemofili A) veya FIX (hemofili B) eksikliği. Etkilenenlerin yaklaşık %30’unda hastalık yeni bir mutasyondan kaynaklanmaktadır.
Epidemiyoloji
Hemofili epidemiyolojisi farklı ülkelerde belirgin farklılıklar gösterse de, hemofiliden şüphelenildiğinde spontan mutasyonların sıklığı nedeniyle, hastaların etnik kökenine büyük önem verilmemektedir. Genel popülasyondaki prevalans 1:12.000 olup, erkek yenidoğanlar arasında görülme sıklığı (1:5.000) önemli ölçüde daha yüksektir. Resesif geçişli bir hastalık olmasına rağmen, heterozigot hamophi lie taşıyan dişilerde hafif semptomlar görülebilir. Pıhtılaşma faktörü eksikliğinin derecesi hastalığın şiddetini belirler.
- Hemofili hemen yalnızca erkeklerde görülen bir kanama eğilimidir.
- Vakaların %85’inde faktör VIII eksikliğine veya anomalisine bağlıdır.Hemofilinin bu tipine hemofili A veya klasik hemofili adı verilir.
- Hemofili hastalarının kalan %15’inde faktör IX eksikliğine bağlı kanama eğilimi görülür.
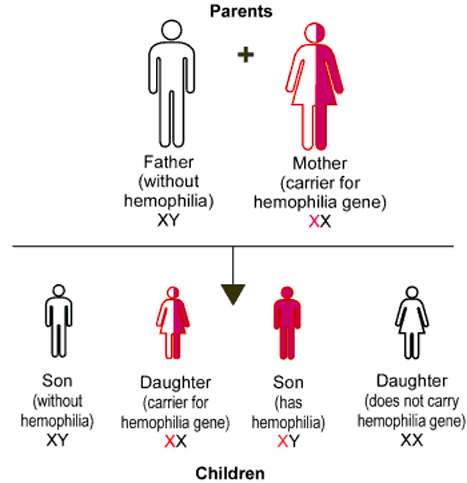
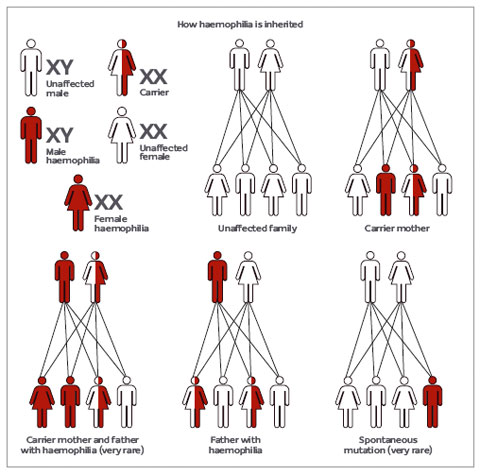
- Her iki faktör de X kromosomlarıyla resesif özellikli genetik geçiş gösterir.bu nedenle, kadınlarda iki kromozomun bu özelliği taşıması gerektiğinden sık görülmez.Bununla birlikte hemofili taşıyıcı olabilirler; erkek çocukların yarısına hastalığı ve kız çocuklarının yarısına da taşıyıcılık özelliğini geçirirler.
- ABD’de 10.000 erkekten 1’inde klasik hemofili vardır.
- Faktör VIII iki komponentten oluşmuştur: molekül ağırlığı milyonlarla ölçülen büyük komponent ve molekül ağırlığı yaklaşık 230.000 olan küçük komponent.küçük komponent pıhtılaşmanın intrensek yolunda çok önemlidir ve faktör VIII’in bu eksikliği klasik hemofiliye neden olur.Farklı karakteristik özellikler taşıyan diğer bir kanama hastalığı olan von Willebrand hastalığı büyük komponentin yokluğuna bağlıdır.
Çeşitleri
Hemofili A, Hemofili B’ye göre 5 kat daha sık görülür.
Patogenez
Hemofilik hastalıkların yaklaşık %30’u spontan bir mutasyondan kaynaklansa da, hastanın aile öyküsünün alınması, laboratuvar testlerinin yanı sıra tanının temel direğidir. Ancak, pozitif bir aile öyküsü dışlama kriteri olarak kullanılmamalıdır. Ek olarak, Von Willebrand sendromunda doğuştan faktör VIII veya faktör IX eksikliğinin olası varlığı göz ardı edilmemelidir, çünkü Von Willebrand pıhtılaşma kaskadının yokluğu, intrinsik pıhtılaşma kaskadının bozulmuş bir süreciyle sonuçlanır. Bu faktörlerin (vWF) yokluğu nedeniyle genellikle F-VIII eksikliği ile faktör X aktive olmayabilir. Von Willebrand faktörü vücutta mevcuttur, böylece daha sonra FVIII’e bağlı daha az trombüs oluşabilir ve bozulmasını önler. Bu da pıhtılaşma faktörünün azalmasına neden olur. Bu durum, iki hastalığın laboratuvar bulguları ve semptomları nedeniyle daha yavaş fibrin oluşumuna ve yara iyileşme bozukluklarına yol açabilir.
Klinik tablo
Faktör eksikliğinin derecesine bağlı olarak, hamofiller başka alt sınıflara ayrılır. 5-40 rezidüel faktör aktivitesine sahip hemofili hafif, %1-5 orta ve <%1 ağır hemofili olarak sınıflandırılır. Ağır hemofilinin ana semptomları genellikle küçük yaralanmalar ve diş çekimi gibi operasyonlardan sonra bile kendiliğinden ve anormal kanamalardır. Orta ve hafif hemofilide, küçük yaralanmalarda da anormal kanamalar meydana gelir, ancak spontan kanama nadirdir. Spontan kanamalar genellikle klinik olarak hemoraji, hematom veya spontan hematüri şeklinde kendini gösterir Deri altı kanamaların genellikle belirli bir komplikasyonu yoktur, ancak tekrarlayan hemorajiler ve kas hematomları genellikle ciddi ikincil hasara neden olur. Etkilenen kişilerde sıklıkla kas atrofileri gelişir. Ayrıca, retrope itoneal kanama tehlikesi her zaman göz önünde bulundurulmalıdır, çünkü düşük semptom seviyesi nedeniyle tanıda bir zorluk teşkil etmektedir. Belirtiler aynı alt sınıflar içinde nispeten değişkendir. Birçok şey hala belirsizdir, ancak hastalığın şiddetinin ilgili gendeki mutasyon türüne de bağlı olduğu kesindir.
Teşhis
Hemofili hastalıklarının yaklaşık %30’u spontan bir mutasyondan kaynaklansa da, şüpheli hastanın aile öyküsünün laboratuvar testleriyle birlikte toplanması, teşhisin temel direğidir. Ancak, negatif bir aile öyküsü asla dışlama kriteri olarak kullanılmamalıdır. Ayrıca, Von Willebrand faktörü (vWF) yokluğu genellikle F-VIII eksikliği ile ilişkili olabileceğinden, Von Willebrand sendromunun olası varlığı göz ardı edilmemelidir. Von Willebrand faktörü plazmada FVIII’e bağlıdır ve bu pıhtılaşma faktörünün parçalanmasını önler. Dolayısıyla, iki hastalığın benzer laboratuvar bulguları ve semptomları nedeniyle belirli sayıda faktörün varlığının doğru tanı koyarken kolayca kafa karışıklığına yol açabilmesidir.
Tedavi
Konvansiyonel tedavi
Hemofili tedavisinde genellikle ihtiyaç duyulan tedavi ile profilaktik tedavi arasında bir ayrım yapılır. İsteğe bağlı tedavi kanama durumunda ve tıbbi müdahalelerden önce kullanılırken, profilaktik tedavi aynı zamanda kas atrofileri veya eklem hastalıkları gibi ağır hemofilinin ikincil hastalıklarını önlemeye yöneliktir. Tedavi, eksik olan pıhtılaşma faktörleri FVII ve FIX’in yerine konmasından oluşur. Profilaktik sürekli tedavide, faktör konsantreleri i.v. olarak 3 kez (hemofili A) veya haftada iki kez (hemofili B) uygulanır, bunun nedeni mevcut preparatların kısa yarı ömrüdür. Yarılanma ömrünün uzatılması, hastalar için haftada gereken uygulama sayısını azaltmak amacıyla şu anda hemofili tedavisindeki temel zorluklardan biridir.
Geleceğin gen terapisi
Diğer bazı monogenetik hastalıklar gibi hemofili de gen tedavisinin kullanıldığı ilk hastalıklardan biridir. Bu, fizyolojik olarak pıhtılaşma faktörleri üretmesi gereken karaciğerin sinüzoidal endotelyal hücrelerinin DNA’sına belirli dizileri yerleştirmeye çalışmak için genetik olarak değiştirilmiş adenovirüslerin kullanılmasını içerir. Ancak, hem yeni üretilen pıhtılaşma faktörlerine hem de genetiği değiştirilmiş karaciğer hücrelerine karşı tepki veren vücudun bağışıklık sistemini atlatmak zordur. Otoimmün reaksiyonlar sıklıkla ortaya çıkar ve immünosupresanlarla kombinasyon tedavisi gerektirir. Devam eden klinik deneyler umut verici sonuçlar göstermektedir. Gen terapisi ile tedavi edilen hastaların büyük bir kısmında en azından şiddetli hemofili – yani <%1 kalıntı faktör aktivitesi – önlenebilmektedir. Bu hastalarda yıllık kanama oranı 8,5 kanama/yıldan 0,3 kanama/yıla düşürülmüştür.