Kardiyomiyopatiler, kalp kasının yapısal ve işlevsel anormallikleriyle karakterize, mekanik veya elektrofizyolojik işlev bozukluğuna yol açan geniş bir miyokardiyal hastalık grubudur. Bu koşullar tipik olarak hipertrofi (kalp kasının kalınlaşması) ile ilişkilidir, ancak hipertrofi tüm vakalarda tanımlayıcı bir özellik değildir. Önemli olarak, kardiyomiyopatiler koroner arter hastalığı (KKH), hipertansiyon, kalp kapak hastalığı veya konjenital kalp defektlerinin neden olduğu kalp hastalıklarından farklıdır.
Tanım ve Terminoloji
Kardiyomiyopatiler, birincil neden olarak koroner arter hastalığının (KKH) bulunmaması ile tanımlanırken, iskemik kardiyomiyopati terimi, kardiyomiyopatinin kesin tanımıyla çelişen doğasına rağmen klinik uygulamada ve literatürde hala sıklıkla kullanılmaktadır. Önemli koroner arter hastalığına ikincil miyokardiyal disfonksiyonu ifade eden İskemik kardiyomiyopati, Amerikan Kalp Derneği (AHA) gibi kuruluşlar tarafından kabul edilmekte ve ICD-10 gibi tanı sistemlerinde yer almaktadır. Dolayısıyla, teknik olarak kardiyomiyopatilerin geleneksel sınıflandırmasının bir parçası olmasa da, iskemik kardiyomiyopati, özellikle dilate kardiyomiyopati bağlamında hala tanınmaktadır.
Tarihsel Sınıflandırma
Kardiyomiyopatiler, Dünya Sağlık Örgütü (DSÖ) tarafından 1995 yılında uzlaşılan bir sınıflandırma sisteminde uzun süre sınıflandırılmıştır. Ancak, genetik katkıların ve yeni tanımlanan kardiyomiyopatilerin anlaşılmasındaki ilerlemelerle birlikte bu sınıflandırma güncelliğini yitirmiştir. Buna karşılık, Moran ve arkadaşları tarafından AHA için geliştirilen gibi güncellenmiş sınıflandırma sistemleri daha incelikli bir çerçeve sağlamıştır.
Kardiyomiyopatilerin Güncel Sınıflandırması
Modern sınıflandırma sistemi kardiyomiyopatileri iki geniş kategoriye ayırır:
- Birincil kardiyomiyopatiler: Öncelikle kalbi etkileyen hastalıklar.
- İkincil kardiyomiyopatiler: Sistemik koşullar veya diğer hastalıklar nedeniyle ortaya çıkan, ancak kalbi daha büyük bir patolojik sürecin parçası olarak içeren hastalıklar.
Primer Kardiyomiyopatiler
Primer kardiyomiyopatiler ayrıca genetik, edinilmiş ve karışık formlara ayrılır:
Genetik Primer Kardiyomiyopatiler
Bu kardiyomiyopatiler kalp kasını, iletim sistemini veya iyon kanallarını etkileyen kalıtsal genetik mutasyonlardan kaynaklanır. Anahtar tipler şunları içerir:
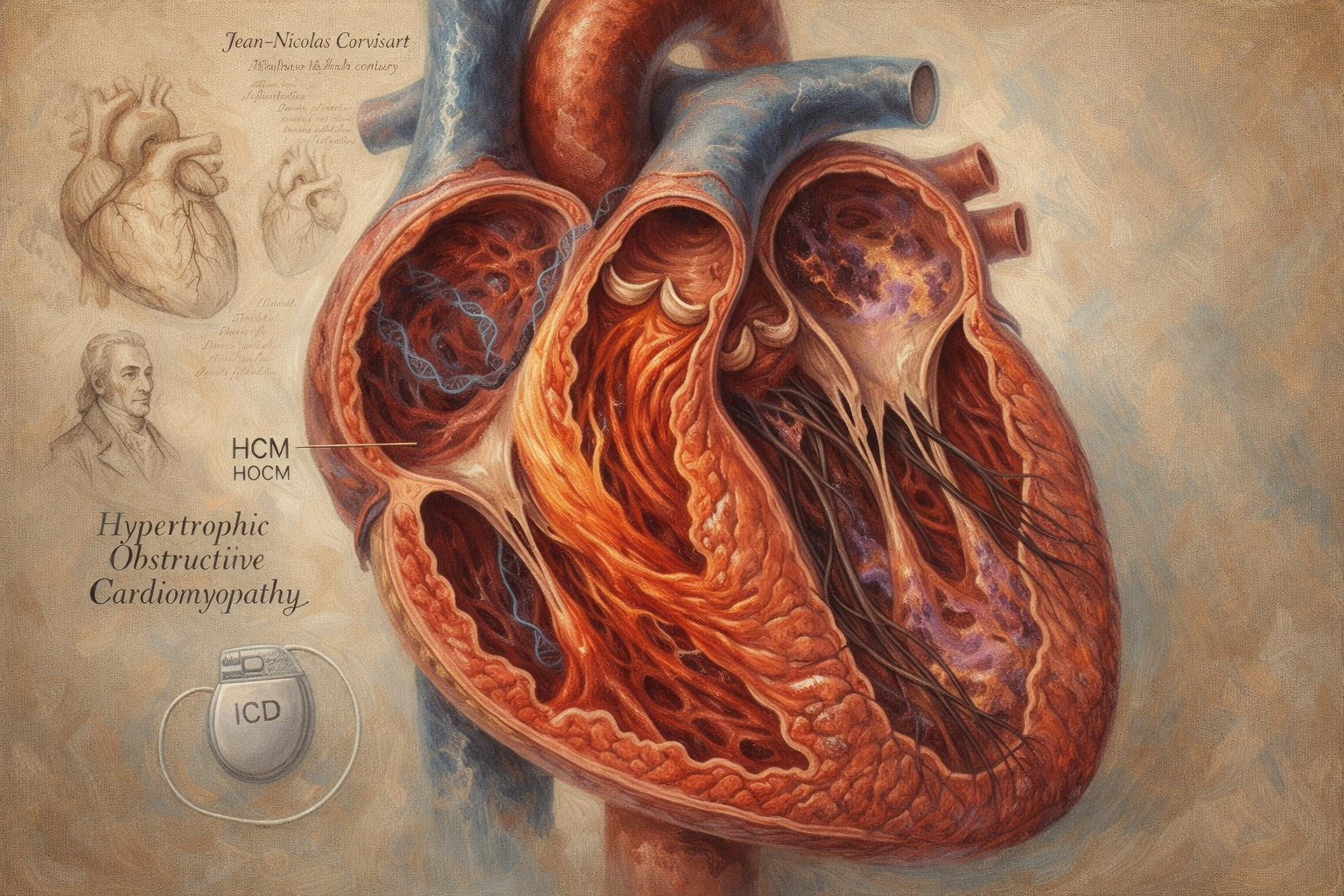
- Ailesel hipertrofik kardiyomiyopati (HCM): Kalp kasının anormal derecede kalınlaştığı ve potansiyel olarak kan akışını engellediği genetik bir bozukluk. Patofizyolojik olarak sıklıkla MYBPC3 gibi sarkomer gen mutasyonlarına bağlı gelişir ve miyofibril düzensizliği (disarray) ile hiperkontraktilite gösterir. Tanıda ilk basamak ekokardiyografidir ve sol ventrikül duvar kalınlığının ≥15 mm olması tanı kriterini karşılar. Kardiyak MR, doku karakterizasyonu ve fibrozis/infiltrasyon değerlendirmesi açısından tamamlayıcıdır; PET-CT ise seçilmiş durumlarda (özellikle eşlik eden inflamatuvar süreçlerde) kullanılır. Tedavide beta-blokerler birinci basamak, kalsiyum kanal blokerleri ikinci seçenektir. Yeni ajanlardan Mavacamten (Camzyos), miyozin inhibisyonu yoluyla hiperkontraktiliteyi azaltır; ancak sol ventrikül ejeksiyon fraksiyonunu (LVEF) düşürebileceğinden dikkatli izlem gerektirir ve gebelikte kontrendikedir.
- Hipertrofik obstrüktif kardiyomiyopati (HOCM): Kalınlaşmış kalp kasının kalpten dışarı kan akışını engellediği bir HCM alt tipidir. Semptomatik obstrüksiyon durumunda cerrahi miyektomi uygulanabilir.
- Klinik Not (HCM/HOCM Komplikasyonları): Atriyal fibrilasyon (AF) bu hasta grubunda sık görülür; esas olarak tromboembolik risk nedeniyle antikoagülasyon gerektirir (kardiyoversiyon öncesinde uygun antikoagülasyon protokolü uygulanmalıdır). Ani kardiyak ölüm (AKÖ) riski AF’den ziyade HCM’ye özgü risk faktörlerine bağlı olup, seçilmiş yüksek riskli hastalarda profilaktik İmplantabl Kardiyoverter Defibrilatör (ICD) endikasyonu doğurur.
- Aritmojenik sağ ventrikül kardiyomiyopatisi (ARVC): Kalbin sağ ventrikülünün yerini yağlı ve fibröz dokunun aldığı ve aritmilere yol açan bir hastalık.
- İletim sistemi kardiyomiyopatileri (Lenegre hastalığı): Bunlar kalbin elektrik iletim sistemindeki anormallikleri içerir.
- İyon kanalı hastalıkları: Kalpteki elektriksel aktiviteyi düzenleyen iyon kanallarını etkileyen mutasyonlardan kaynaklanan long-QT sendromu, kısa-QT sendromu, Brugada sendromu ve katekolaminerjik polimorfik ventriküler taşikardi (CPVT) gibi bozukluklar.
Karma Genetik-Kazanılmış Kardiyomiyopatiler
Bu kardiyomiyopatiler hem genetik yatkınlık hem de edinilmiş faktörlerin özelliklerini sergiler:
- Dilate kardiyomiyopati (DCM): Kanı etkili bir şekilde pompalayamayan genişlemiş bir kalp ile karakterize edilir. Bu durum genetik olabilir, ancak enfeksiyonlar, toksinler veya otoimmün bozukluklardan da kaynaklanabilir.
- Restriktif kardiyomiyopati (RCM): Kalp kasının sertleşerek diyastol (gevşeme fazı) sırasında kalbin dolmasını kısıtladığı nadir bir kardiyomiyopati türüdür. Hipertrofik kardiyomiyopatilerin aksine, RCM genellikle kas kalınlaşmasını içermez.
Kazanılmış Primer Kardiyomiyopatiler
Bu koşullar genetik yatkınlık olmaksızın çevresel faktörlerden veya fizyolojik stresten kaynaklanır:
- Miyokardit ile ilişkili kardiyomiyopati: Genellikle enfeksiyonlara bağlı olarak kalp kasının iltihaplanması kardiyomiyopatiye yol açabilir.
- Tako-Tsubo kardiyomiyopatisi (Stres kaynaklı veya “kırık kalp” sendromu): Tipik olarak şiddetli duygusal veya fiziksel stresle tetiklenen, kalp krizini taklit eden ancak koroner arter tıkanıklığı olmayan geçici bir kardiyomiyopati.
- Peripartum kardiyomiyopati: Hamileliğin son ayında veya doğum sonrası beş ay içinde ortaya çıkan nadir bir kalp yetmezliği şekli.
Sekonder Kardiyomiyopatiler
İkincil kardiyomiyopatiler, infiltratif, toksik, metabolik, enflamatuar veya otoimmün bozukluklar gibi kalbi tutan sistemik hastalıklar veya durumlardan kaynaklanır.
İnfiltratif Kardiyomiyopatiler
Bu koşullar, maddelerin kalp kası içine anormal şekilde birikmesini veya sızmasını içerir:
- Amiloidoz: Amiloid proteinlerinin birikmesi kalbi etkileyerek kısıtlayıcı kardiyomiyopatiye yol açabilir. Özellikle Transtiretin (TTR) amiloidozu, HCM ile benzer şekilde sol ventrikül duvar kalınlaşması ve belirgin diyastolik disfonksiyonla seyreden temel bir patolojidir; ancak patofizyolojisi tamamen farklıdır (miyokard içine protein birikimi sonucu restriktif bir doluş paterni ortaya çıkar). Bu iki hastalığın ayırıcı tanısında EKG’de düşük voltaj ile ekokardiyografide kalın duvar görünümü arasındaki uyumsuzluk TTR amiloidozu için tipiktir. Klinik olarak karpal tünel sendromu gibi erken sistemik bulgular özellikle TTR amiloidozunu düşündürür. HCM’nin aksine, amiloidozda beta-bloker toleransı sınırlı olabilir. Bu grupta da atriyal fibrilasyon gelişimi sıktır ve tromboembolik risk nedeniyle antikoagülasyon şarttır.
- Gaucher hastalığı, Hurler sendromu, Hunter sendromu: Kalbe sızabilen ve kardiyomiyopatiye neden olabilen lizozomal depolama bozuklukları.
- Hemokromatoz: Kalp kasına zarar veren bir aşırı demir yüklenmesi durumu.
- Fabry hastalığı, Pompe hastalığı, Niemann-Pick hastalığı: Belirli lipidlerin veya glikojenin depolanmasını etkileyen, genellikle kalbi ilgilendiren genetik bozukluklar.
Toksik Nedenler
Kalbe zarar verebilecek ve kardiyomiyopatiye yol açabilecek maddeler şunlardır:
- Uyuşturucular: Kokain ve bazı kemoterapi ilaçları (örn. antrasiklinler, siklofosfamid).
- Ağır metaller: Kadmiyum ve diğer çevresel toksinler toksik kardiyomiyopatiye yol açabilir.
Endokrinolojik Nedenler
Çeşitli endokrin bozukluklar ikincil kardiyomiyopatilere neden olabilir:
- Diabetes mellitus: Kronik yüksek kan şekeri diyabetik kardiyomiyopatiye yol açabilir.
- Hipertiroidizm ve hipotiroidizm: Hem aşırı aktif hem de az aktif tiroid bezleri kalbin işlevini etkileyebilir.
- Hiperparatiroidizm, feokromositoma, akromegali: Bu endokrin bozukluklar hormon seviyelerini etkileyerek dolaylı olarak kalp kası hasarına yol açar.
İnflamatuar Nedenler
Kalp kasını etkileyen iltihaplı hastalıklar şunlardır:
- Löffler endokarditi: Kalbin eozinofilik infiltrasyonu ile karakterize olup kısıtlayıcı kardiyomiyopatiye yol açar.
- Endomiyokardiyal fibrozis: Fonksiyonu kısıtlayan kalp iç zarının fibrozisi.
- Sarkoidoz: Kalpte granülomlara neden olan, potansiyel olarak aritmilere ve kalp yetmezliğine yol açan bir hastalık.
Nörolojik Nedenler
Bazı nörolojik hastalıklar kardiyomiyopati ile ilişkilidir, örneğin:
- Kas distrofisi (Becker, Duchenne): Kalp kası da dahil olmak üzere kas bütünlüğünü etkileyen genetik bozukluklar.
- Friedreich ataksisi: Sinir sistemi ve kalpte ilerleyici hasara neden olan nadir bir genetik hastalık.
- Nörofibromatozis: Sinir hücrelerinin gelişimini etkileyen, tümörlere ve bazen kalp tutulumuna yol açan genetik bir bozukluk.
Otoimmün Nedenler
Aşağıdakiler de dahil olmak üzere çeşitli otoimmün hastalıklar kardiyomiyopatiye yol açabilir:
- Sistemik lupus eritematozus (SLE): Lupustan kaynaklanan kronik enflamasyon kalbi etkileyebilir.
- Romatoid artrit, skleroderma, dermatomiyozit ve polyarteritis nodosa: Bu otoimmün durumlar kalp kasını iltihaplandırabilir veya yaralayabilir.
Besinsel Nedenler
Ciddi beslenme eksiklikleri de kardiyomiyopati ile ilişkilendirilmektedir:
- Hipovitaminoz (Beri-beri, Pellagra, Scurvy): Temel vitaminlerdeki (tiamin, niasin ve C vitamini gibi) eksiklikler kalp yetmezliğine neden olabilir.
- Selenyum eksikliği: Düşük selenyum seviyeleri kardiyomiyopatiye neden olabilir.
- Kwashiorkor: Tipik olarak şiddetli yetersiz beslenmede görülen protein eksikliği kalbe zarar verebilir.
- Karnitin eksikliği: Yağ asidi metabolizması için gerekli bir besin maddesi olan karnitin eksikliği kalp fonksiyonlarını bozabilir.
Diğer Nedenler
Kardiyomiyopatilerin diğer ikincil nedenleri şunlardır:
- Noonan sendromu: Kalbi etkileyebilen genetik bir bozukluk.
- Kemoterapi: Kardiyomiyopati, antrasiklinler ve siklofosfamid gibi kemoterapötik ajanların kullanımından kaynaklanabilir.
- Radyoterapi: Özellikle göğüs bölgesine uygulanan radyasyon tedavisi zamanla kalp hasarına neden olabilir.
Tedavi Prensipleri
HCM Tedavisi:
- Semptomatik hastalarda beta-blokerler (propranolol, metoprolol) birinci basamaktır; non-dihidropiridin kalsiyum kanal blokerleri (verapamil) ikinci seçenek.
- Mavacamten (Camzyos): Kardiyak miyozin inhibitörü; hiperkontraktiliteyi doğrudan azaltır, LVOT gradientini düşürür. LVEF’yi azaltabileceği için düzenli ekokardiyografik izlem şarttır. Gebelikte kontrendikedir.
- Obstrüktif HCM’de septa miyektomi veya alkol septal ablasyon.
- Ani kardiyak ölüm (SCD) riski yüksekse (kalınlık ≥30 mm, ailede SCD öyküsü, non-sustained VT, anormal kan basıncı yanıtı vb.) profilaktik ICD endikasyonu doğar.
ATTR Amiloidoz Tedavisi:
- Beta-blokerler genellikle kötü tolere edilir (bradikardi, düşük debi nedeniyle).
- Tafamidis (TTR stabilizatörü) hastalığın progresyonunu yavaşlatır.
- Destek tedavisi: diüretikler (dikkatli volüm yönetimi), AF kontrolü.
Ortak Yaklaşımlar:
- Her iki durumda da atriyal fibrilasyon çok sıktır ve CHA₂DS₂-VASc skoru ne olursa olsun antikoagülasyon (DOAK veya varfarin) endikedir.
- Kardiyoversiyon öncesi en az 3-4 hafta antikoagülasyon ve TEE ile sol atrium trombüs dışlanması önerilir.
- Kalp yetmezliği tedavisi: SGLT2 inhibitörleri, ARNI/ACEi/ARB (amiloidozda hipotansiyon riski nedeniyle dikkatli), mineralokortikoid reseptör antagonistleri.
Prognoz ve İzlem
HCM’de prognoz genetik varyanta, duvar kalınlığına, obstrüksiyona ve aritmiye bağlıdır. ATTR amiloidozda erken tanı (özellikle tafamidis başlanması) sağkalımı belirgin iyileştirir. Her iki hastalıkta da düzenli EKO, Holter, kardiyak MR ve multidisipliner (kardiyolog, genetikçi, hematolog) takip esastır.
Bu geniş çerçeve, kardiyomiyopatilerin hem genetik/moleküler temellerini hem de klinik pratikte en sık karşılaşılan ayırıcı tanılardan HCM ve kardiyak amiloidozu bütüncül bir yaklaşımla ele almaktadır. Güncel kılavuzlar (ESC 2023 HCM, ATTR Amyloidosis consensus) bu ayrımları ve tedavi algoritmalarını daha da detaylandırmaktadır.
Keşif
Temel olarak miyokardı etkileyen bir grup kalp hastalığı olan kardiyomiyopatiler, önemli bilimsel atılımlarla işaretlenmiş zengin ve gelişen bir geçmişe sahiptir. Bu dönüm noktaları, bu esrarengiz durumları anlamak ve tedavi etmek için çalışan bilim insanlarının ve klinisyenlerin ısrarlı merakını ve kararlılığını vurgulamaktadır.
Erken Gözlemler (18. ve 19. Yüzyıllar)
Kalp kası hastalıkları kavramı ancak 18. yüzyılın sonlarında ve 19. yüzyılın başlarında, doktorların kalp kasındaki anormalliklerden kaynaklanan kalp yetmezliğini kapak veya koroner hastalıklardan kaynaklananlardan ayırt etmeye başlamasıyla ortaya çıkmaya başlamıştır. Napolyon Bonapart’ın kişisel doktoru olarak görev yapan Fransız hekim Jean-Nicolas Corvisart miyokardiyal anormalliklerin tanımlanmasında ilk öncülerden biriydi. Otopsi raporlarında, kalp yetmezliğinden ölen hastalarda genişlemiş veya kalınlaşmış kalp kasları tanımlayarak, kalp hastalığının herhangi bir belirgin kapak sorunu veya koroner tıkanıklık olmadan da ortaya çıkabileceğini öne sürdü. Bu gözlemlerin ayrı bir isim altında kategorize edilmesi uzun yıllar alacak olsa da, Corvisart’ın çalışmaları daha sonra kardiyomiyopatiler olarak tanınacak olan hastalıklar için anlayış tohumlarını ekti.
1957: “Kardiyomiyopati” Teriminin Kullanılmaya Başlanması
Önemli bir dönüm noktası 1957 yılında Wallace Brigden tarafından “kardiyomiyopati ” teriminin ortaya atılmasıyla yaşandı. İngiliz bir doktor olan Brigden, koroner arter hastalığı, kapak hastalığı veya hipertansiyon ile açıklanamayan kalp kası hastalıklarını sınıflandıran ilk kişiler arasındaydı. Çalışmaları, kardiyomiyopatilerin miyokardı etkileyen ayrı bir hastalık grubu olarak ayırt edilmesine zemin hazırladı. Bu, kalp tıbbında ileriye doğru atılan muazzam bir adımdı çünkü bu noktaya kadar kalp hastalıklarının çoğu öncelikle koroner arter hastalığı merceğinden görülüyordu.
Brigden konseptini tanıttığında, koroner arter hastalığına odaklanmış olan ve tıkalı arterlerle ilgisi olmayan miyokardiyal hastalıkların varlığını kabul etmekte zorlanan birçok meslektaşının şüpheciliğiyle karşılaştı. Ancak zamanla, daha fazla açıklanamayan kalp kası hastalığı vakası bildirildikçe teorisi ilgi gördü ve “kardiyomiyopati” terimi tıp literatüründe yaygın olarak kabul görmeye başladı. Bu, bu koşulların doğası ve nedenlerine ilişkin gelecekteki araştırmalara zemin hazırladığı için önemli bir dönüm noktasıydı.
1980s: Genetik Kardiyomiyopatilerin Keşfi
1980’lerde kardiyomiyopatiler yoğun genetik araştırmaların yapıldığı bir alan haline gelmiştir. Bu dönemde araştırmacılar ailesel paternleri tanımlamaya başladılar ve bu da birçok kardiyomiyopatinin kalıtsal olduğunun keşfedilmesine yol açtı. Genetik mutasyonlar, özellikle hipertrofik kardiyomiyopati (HCM) olmak üzere belirli kardiyomiyopati türlerinin altında yatan neden olarak belirlendi. Bu keşif, kardiyomiyopatilerin nasıl geliştiği ve genetiğin hastalıkta oynadığı rolün anlaşılmasında devrim yarattı.
Hipertrofik kardiyomiyopati tarihindeki en dokunaklı anlardan biri 1990 yılında, önde gelen bir kolej basketbol oyuncusu olan Hank Gathers bir maç sırasında sahada yere yığılıp öldüğünde yaşandı. Gathers’a kalp kasının kalınlaşmasına neden olarak kalbin kan pompalamasını zorlaştıran genetik bir durum olan HCM teşhisi konmuştu. Ölümü, özellikle genç sporcularda teşhis edilmemiş veya kötü yönetilen kardiyomiyopatilerle ilişkili risklerin altını çizdi. Bu trajik olay, genetik kalp hastalıkları için daha iyi tarama yapılması ihtiyacına dünya çapında dikkat çekmiş ve hipertrofik kardiyomiyopati konusunda kamuoyunda yaygın bir farkındalık yaratmıştır.
1995: DSÖ Sınıflandırma Sistemi
1995 yılında Dünya Sağlık Örgütü (WHO) kardiyomiyopatiler için resmi bir sınıflandırma sistemi geliştirerek tıp alanındaki yerlerini farklı hastalıklar olarak daha da sağlamlaştırmıştır. Sınıflandırma sistemi kardiyomiyopatileri üç ana tipe ayırmıştır: Dilate, hipertrofik ve restriktif kardiyomiyopati. Bu yapılandırılmış sınıflandırma, klinisyenlerin bu durumları teşhis etmesini ve yönetmesini kolaylaştırdı ve nedenleri ve tedavileri hakkında daha fazla araştırma yapılması için bir temel sağladı.
Aynı zamanda, özellikle yüksek profilli dilate kardiyomiyopati (DCM) vakaları daha yaygın olarak bildirilmeye başlandıkça, halk bu hastalıkları daha fazla fark etmeye başladı. Bu vakalardan biri Kuzey İrlandalı efsanevi futbolcu George Best ile ilgiliydi. Saha içindeki olağanüstü yetenekleri ve saha dışındaki çalkantılı yaşamıyla tanınan Best’e, hayatının ilerleyen dönemlerinde, büyük ölçüde alkol bağımlılığının bir sonucu olarak DCM teşhisi kondu. Erken ölümüne neden olan bu hastalıkla mücadelesi, alkol tüketimi gibi yaşam tarzı faktörlerinin kardiyomiyopatilerin gelişimi üzerindeki etkisine ışık tutmuştur.
2000’ler: Genetik Test ve Tedavideki Gelişmeler
2000’li yıllar, kardiyomiyopatilerin daha erken ve daha doğru bir şekilde teşhis edilmesine olanak tanıyan yeni bir genetik test çağını başlattı. Moleküler biyolojideki gelişmeler, hipertrofik ve dilate kardiyomiyopati gibi durumlardan sorumlu mutasyonların belirlenmesini mümkün kıldı. Risk altındaki aileler için genetik tarama daha yaygın hale gelerek erken müdahalelere ve hastalığın daha iyi yönetilmesine olanak sağladı. Bu dönemde ayrıca implante edilebilir kardiyoverter defibrilatörler (ICD’ler) ve sol ventrikül destek cihazları (LVAD’ler) geliştirilerek kardiyomiyopatinin ağır formlarına sahip hastaların hayatta kalma oranları iyileştirildi.
Bu gelişmelerden faydalanan en ünlü isimlerden biri, 2012 yılında kendisine kardiyomiyopati geliştirmesine neden olan bir durum olan lupus nefriti teşhisi konulduğunu açıklayan Amerikalı televizyon sunucusu ve aktör Nick Cannon olmuştur. Teşhisi ve kalp ameliyatı ve yardımcı cihazların kullanımını içeren tedavisi hakkındaki açıklığı, kardiyomiyopatiler hakkında farkındalık yaratmaya yardımcı oldu ve başkalarını uygun bakım aramaya teşvik etti.
Günümüz: Kişiselleştirilmiş Tıp ve Kardiyomiyopati
Günümüzde kardiyomiyopatiler üzerine yapılan çalışmalar, kişiye özel tıbba odaklanarak gelişmeye devam etmektedir. Genetik araştırmalar, klinisyenlerin tedavileri hastalarda bulunan spesifik genetik mutasyonlara göre uyarlamasını sağlamıştır. Bireyselleştirilmiş bakıma yönelik bu değişim, hastalığın ilerlemesini daha iyi tahmin etmeyi ve komplikasyon riskini azaltan hedefe yönelik tedaviler sunmayı mümkün kılmıştır.
2019 yılında spor dünyasından dikkat çekici bir hikaye ortaya çıktı: Uluslararası üne sahip futbol yıldızı Cristiano Ronaldo, gençken kardiyomiyopati gelişimine yol açabilecek bir durum olan taşikardi teşhisi konduğunda kariyerinin neredeyse başlamadan sona erdiğini açıkladı. Sorunu düzeltmek için bir kalp ameliyatı geçiren Ronaldo, tarihteki en büyük sporculardan biri olmaya devam etti. Ronaldo’nun hikayesi, potansiyel olarak yaşamı tehdit eden durumların ilerlemesini önleyebilecek erken teşhis ve müdahalenin önemini vurgulamıştır.
Kalp kası anormalliklerinin ilk gözlemlerinden genetik mutasyonların keşfine ve modern tedavilerin geliştirilmesine kadar kardiyomiyopatilerin tarihi, bilimsel araştırma ve yeniliğin gücünün bir kanıtıdır. Her bir dönüm noktası bizi bu karmaşık hastalıkları anlamaya ve etkilenenlerin yaşamlarını iyileştirmeye daha da yaklaştırmıştır.
İleri Okuma
- Towbin, J. A., & Bowles, N. E. (2002). “The failing heart.” Nature, 415(6868), 227-233.
- Elliott, P., & McKenna, W. J. (2004). “Hypertrophic cardiomyopathy.” The Lancet, 363(9424), 1881-1891.
- Maron, B. J., Towbin, J. A., Thiene, G., Antzelevitch, C., Corrado, D., Arnett, D., … & Siscovick, D. (2006). “Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement.” Circulation, 113(14), 1807-1816.
- Elliott, P., Andersson, B., Arbustini, E., Bilinska, Z., Cecchi, F., Charron, P., … & McKenna, W. J. (2008). “Classification of the cardiomyopathies: a position statement from the European Society of Cardiology.” European Heart Journal, 29(2), 270-276.
- Morita, H., Rehm, H. L., Menesses, A., McDonough, B., Roberts, A. E., Kucherlapati, R., … & Seidman, J. G. (2008). “Shared genetic causes of cardiac hypertrophy in children and adults.” New England Journal of Medicine, 358(18), 1899-1908.