Tanım
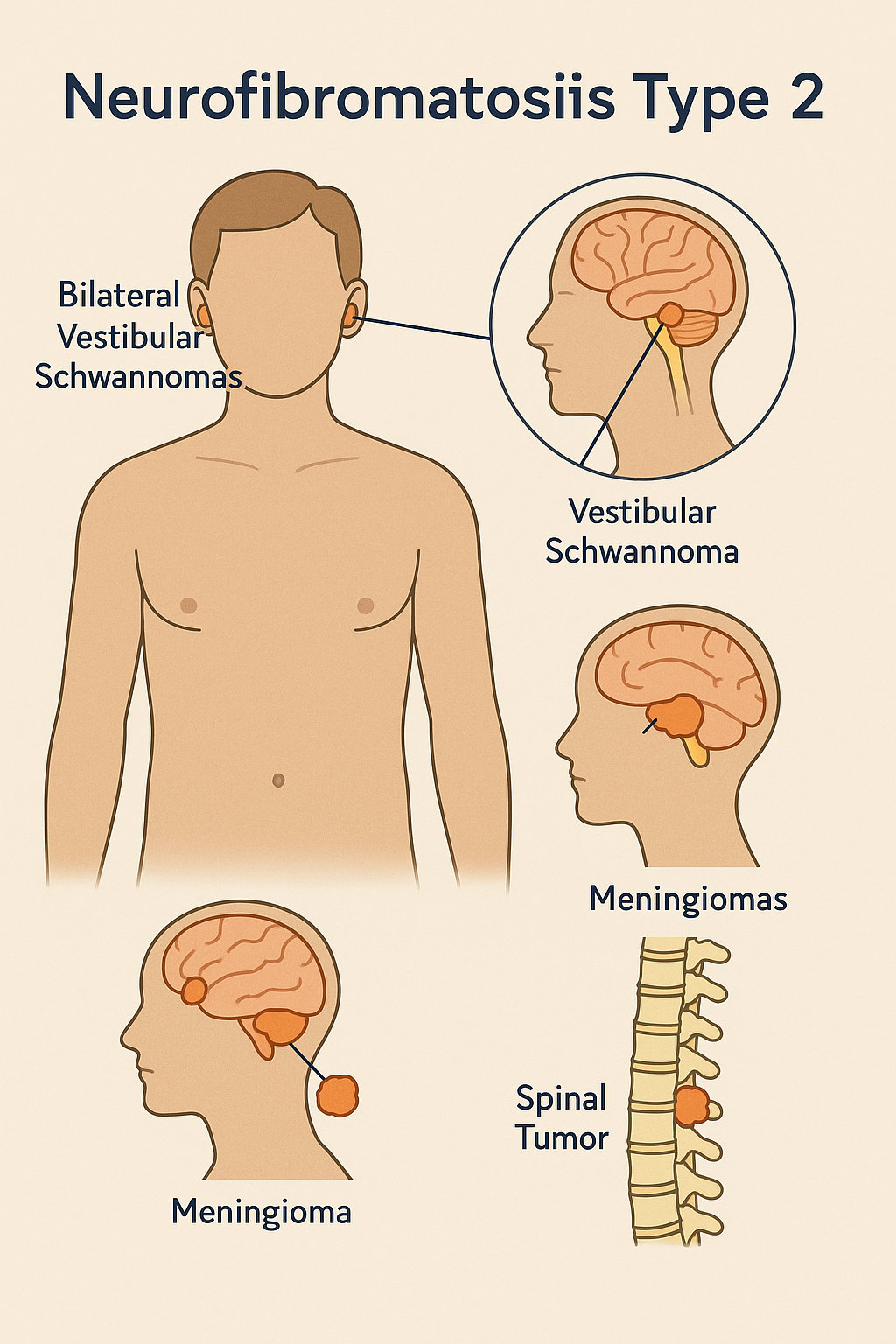
NF2, NF1 ve schwannomatosis’ten farklı olarak nörofibromatozis grubunda otozomal dominant kalıtsal bir tümör sendromudur. İyi huylu sinir sistemi tümörleri, özellikle bilateral vestibüler schwannomlar ile karakterizedir.
Genetik
- Gen: 22q12 kromozomunda bulunan ve hücre zarı stabilitesi ve büyüme sinyalizasyonu için kritik bir tümör baskılayıcı olan Merlin’i (schwannomin) kodlayan NF2 geni.
- Kalıtım: Otozomal dominant, ancak vakaların yaklaşık %50’si de novo mutasyonlardan kaynaklanır.
- Patogenez: Merlin kaybı, kontrolsüz Schwann hücresi ve meningeal hücre çoğalmasına yol açarak schwannomlara ve menenjiyomlara neden olur.
Epidemiyoloji
- İnsidans: 25.000-30.000 kişide ~1.
- Başlangıç: Semptomlar genellikle geç ergenlik/erken yetişkinlik döneminde (18-24 yaş) ortaya çıkar, ancak asemptomatik vakalar daha erken görülebilir.
Klinik Özellikler
Belirgin Belirtiler:
- İki taraflı vestibüler schwannomlar (akustik nörinomlar): Hastaların >%90’ında görülür; işitme kaybına, kulak çınlamasına ve denge sorunlarına neden olur.
- Spinal/Periferik schwannomlar ve meningiomlar (intrakranial/spinal).
- Ependimomlar (omurilik tümörleri) – sorguda muhtemelen “elektrifikasyon” olarak anılmıştır (muhtemelen bir yazım hatasıdır).
Oküler Anormallikler:
- Posterior subkapsüler kataraktlar (hastaların %80’ine kadar).
- Retinal hamartomlar.
Kutanöz Özellikler:
- Café-au-lait lekeleri (NF1’den daha az) ve deri schwannomaları (plak benzeri veya deri altı).
Nörolojik Komplikasyonlar:
- Polinevropati (tümör kompresyonu veya doğrudan sinir tutulumu nedeniyle).
- Kranial sinir felçleri (örn. yüz zayıflığı).
Tanı Kriterleri
- Kesin NF2:
- İki taraflı vestibüler schwannomlar VEYA
- NF2 aile öyküsü + tek taraflı vestibüler schwannom VEYA
- İki NF2 ile ilişkili tümör (örn. menenjiyom, schwannom, ependimom) + tek taraflı vestibüler schwannom.
- Genetik test kan/tümör dokusunda NF2 mutasyonlarını doğrular.
Ayırıcı Tanı
- NF1: Kromozom 17, nörofibromlar, Lisch nodülleri, kemik lezyonları.
- Schwannomatozis: Çoklu schwannomlar (vestibüler hariç), SMARCB1/LZTR1 mutasyonları.
Yönetim
Cerrahi/Girişimsel:
- Semptomatik rahatlama için Tümör rezeksiyonu (örn. işitme kaybı, beyin sapı basısı).
- İşitme koruma stratejileri: İşitsel beyin sapı implantları/koklear implantlar.
- Radyasyon tedavisi (tartışmalı; kötü huylu dönüşüm riski).
Tıbbi Tedaviler:
- Bevacizumab (anti-VEGF): Vestibüler schwannom boyutunu küçültür ve işitmeyi stabilize eder.
- Lapatinib (EGFR/HER2 inhibitörü): Erken denemelerde potansiyel göstermektedir.
Destekleyici Bakım:
- Düzenli MRI gözetimi (beyin/omurga).
- Denge sorunları için fizik tedavi.
- Genetik danışmanlık ve aile taraması.
Prognoz
- Değişken; tümör yüküne ve komplikasyonlara (örneğin, işitme kaybı, omurilik basısı) bağlıdır.
- Beyin sapı basısı veya kötü huylu ilerleme nedeniyle şiddetli vakalarda yaşam süresinin azalması (nadir).
Önemli Çıkarım: NF2, erken tanı, genetik danışmanlık ve işlevi korumak için özel müdahalelere vurgu yapan ömür boyu multidisipliner bakım gerektirir.
Keşif
Erken Klinik Gözlemler
NF2 ile ilişkili bir tümör tipinin en erken kaydedilen sözü, Eduard Sandifort’un bir otopsi sırasında akustik nöromayı (şimdi vestibüler schwannoma olarak bilinir) tanımladığı 1777 yılına dayanır Nörofibromatozis tip 2’nin erken tarihi. Bu, henüz özel olarak NF2 ile ilişkilendirilmemiş olsa da önemli bir erken gözlemdi. 1811’de Louis Older, sinir neoplazmalarını tanımlamak için “nöroma” terimini türeterek daha sonraki sınıflandırmalar için temel bir terim oluşturdu Nörofibromatozis tip 2’nin erken tarihi.
1822’de John H. Wishart, bilateral akustik nöromlar ve menenjiyomlar sergileyen 21 yaşındaki kalfa fırıncı Michael Blair’in ayrıntılı bir vakasını sunduğunda önemli bir an yaşandı (Nörofibromatozis tip 2’nin erken tarihi). Bu vaka artık NF2’nin ilk net klinik tanımı olarak kabul ediliyor ve bu tümörleri durumla ilişkilendiriyor. Wishart’ın çalışması, erken, agresif NF2 sunumları için “Wishart tipi” sınıflandırmasına yol açtı. 19. yüzyıldaki diğer katkılar arasında Jean Cruveilhier’in 1835’te akustik nöromaların intrakraniyal etkilerine ilişkin analizi ve Knoblauch’un 1843’te bilateral akustik nöromalara ilişkin raporu yer aldı ve giderek artan kanıtlara Nörofibromatozis tip 2’nin erken tarihi eklendi.
NF2’yi NF1’den ayırt etme
NF2 ile Nörofibromatozis Tip 1 (NF1) arasındaki ayrım 19. yüzyılın sonlarında ve 20. yüzyılın başlarında ortaya çıkmaya başladı. 1897’de Mossé ve Cavalié, periferik belirtileri olmayan nörofibromatozis varyantlarını tanımlamak için “santral nörofibromatozis” terimini ortaya attılar ve diğer kranial sinir nöromalarıyla birlikte bilateral akustik nöromaları da tanıdılar Nörofibromatozis tip 2’nin erken tarihi. Bu, NF2’yi farklılaştırmaya yönelik önemli bir adımdı. 1903’te Henneberg ve Koch, bilateral 8. kranial sinirleri (vestibülokoklear sinirler) içeren belirgin bir intrakranial formu tanımlayarak bunu daha da açıklığa kavuşturdular ancak cildi koruyarak “merkezi” ve “periferik” nörofibromatozis kavramını tanıttılar Nörofibromatozis tip 2’nin erken tarihi. Bu ayrım, NF2’nin ayrı bir varlık olarak tanınması için hayati önem taşıyordu.
Ailesel Modeller ve Kalıtım
- yüzyılın ortalarına doğru, raporlar NF2’nin ailesel oluşumunu vurgulamaya başladı ve otozomal dominant bir kalıtım modelini önerdi. Dikkat çekici bir örnek, Gardiner ve Frazier’in beş nesil boyunca 38 etkilenen üyeye sahip bir aile raporuydu; eksik penetrans veya cinsiyet özgüllüğü olmadan %50 kalıtım riski belirtiliyordu Nörofibromatozis tip 2’nin erken öyküsü. 1987’de Young ve arkadaşları bunu genişleterek dokuz nesil boyunca aynı ailenin 97 üyesini incelediler ve bu da NF2’nin ayrı bir genetik bozukluk olduğunu belirlemeye yardımcı oldu Nörofibromatozis tip 2’nin erken öyküsü. Bu ailenin daha hafif seyri, daha agresif “Wishart tipi” ile tezat oluşturan NF2 için “Gardner alt tipi” teriminin kullanılmasına yol açtı.
Resmi Tanıma ve Genetik İçgörüler
NF1 ve NF2’nin resmi farklılaşması, 1987’de Ulusal Sağlık Enstitüleri (NIH) konsensüs panelinin bunların farklı genetik bozukluklar olduğunu doğrulamasıyla gerçekleşti ve bu, NF2’nin tanınmasında önemli bir dönüm noktası oldu Nörofibromatozis tip 2’nin erken tarihi. Bunu 20. yüzyılın sonlarında genetik atılımlar izledi. 1986’da Seizinger ve arkadaşları, insan akustik nöromalarının tümör oluşumunda kromozom 22’deki genlerin kaybını bildirerek NF2’yi bu kromozomal bölgeye bağladı Nörofibromatozis: kronolojik tarih ve güncel konular. 1990 yılında, Rouleau ve arkadaşları NF2 genini 22. kromozomun uzun kolunda (q12) buldular ve bunun bir tümör baskılayıcı gen gibi davrandığını öne sürdüler Nörofibromatozis: kronolojik tarih ve güncel konular. 1993 yılında, NF2 geni Rouleau ve arkadaşları tarafından klonlandı ve Trofatter ve arkadaşları bunun moesin, ezrin ve radixin’e benzer, günümüzde merlin (veya schwannomin) olarak bilinen bir proteini kodladığını tespit ettiler Nörofibromatozis: kronolojik tarih ve güncel konular. Bu protein hücre şekli ve hareketinde kritik bir rol oynar ve işlev bozukluğu NF2’deki tümör oluşumuyla bağlantılıdır.
İleri Okuma
- Wishart, J. (1822). Case of tumours in the dura mater. Edinburgh Medical and Surgical Journal, 18, 393–397.
- von Recklinghausen, F. D. (1882). Über die multiplen Fibrome der Haut und ihre Beziehung zu den multiplen Neuromen. Berlin: A. Hirschwald.
- Feiling, A. (1920). Bilateral acoustic tumors. Lancet, 196(2), 1072.
- Gardner, W. J., & Frazier, C. H. (1930). Central neurofibromatosis: A neuropathological study of two cases. Archives of Neurology and Psychiatry, 23(2), 211–225.
- Crowe, F. W., Schull, W. J., & Neel, J. V. (1956). A clinical, pathological, and genetic study of multiple neurofibromatosis. Springfield, IL: Charles C. Thomas.
- Riccardi, V. M. (1982). Neurofibromatosis: Phenotype, Natural History, and Pathogenesis. Johns Hopkins University Press.
- Riccardi, V. M. (1987). Von Recklinghausen neurofibromatosis. New England Journal of Medicine, 317(13), 883–889.
- Rouleau, G. A., Wertelecki, W., Haines, J. L., et al. (1987). Genetic linkage of bilateral acoustic neurofibromatosis to a DNA marker on chromosome 22. Nature, 329(6141), 246–248.
- Seizinger, B. R., Rouleau, G. A., Ozelius, L. J., Lane, A. H., Huson, S. M., Wallace, M. R., … & Gusella, J. F. (1987). Genetic linkage of von Recklinghausen neurofibromatosis to the centromeric region of chromosome 17. Nature, 329(6141), 289–291.
- Wallace, M. R., Marchuk, D. A., Andersen, L. B., et al. (1990). Type 2 neurofibromatosis gene: Identification of a chromosome 22 gene with multiple exons mutated in NF2. Science, 249(4965), 181–186.
- Trofatter, J. A., MacCollin, M. M., Rutter, J. L., et al. (1993). A novel moesin-ezrin-radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell, 72(5), 791–800.
- Rouleau, G. A., Merel, P., Lutchman, M., et al. (1993). Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature, 363(6429), 515–521.