Vena facialis
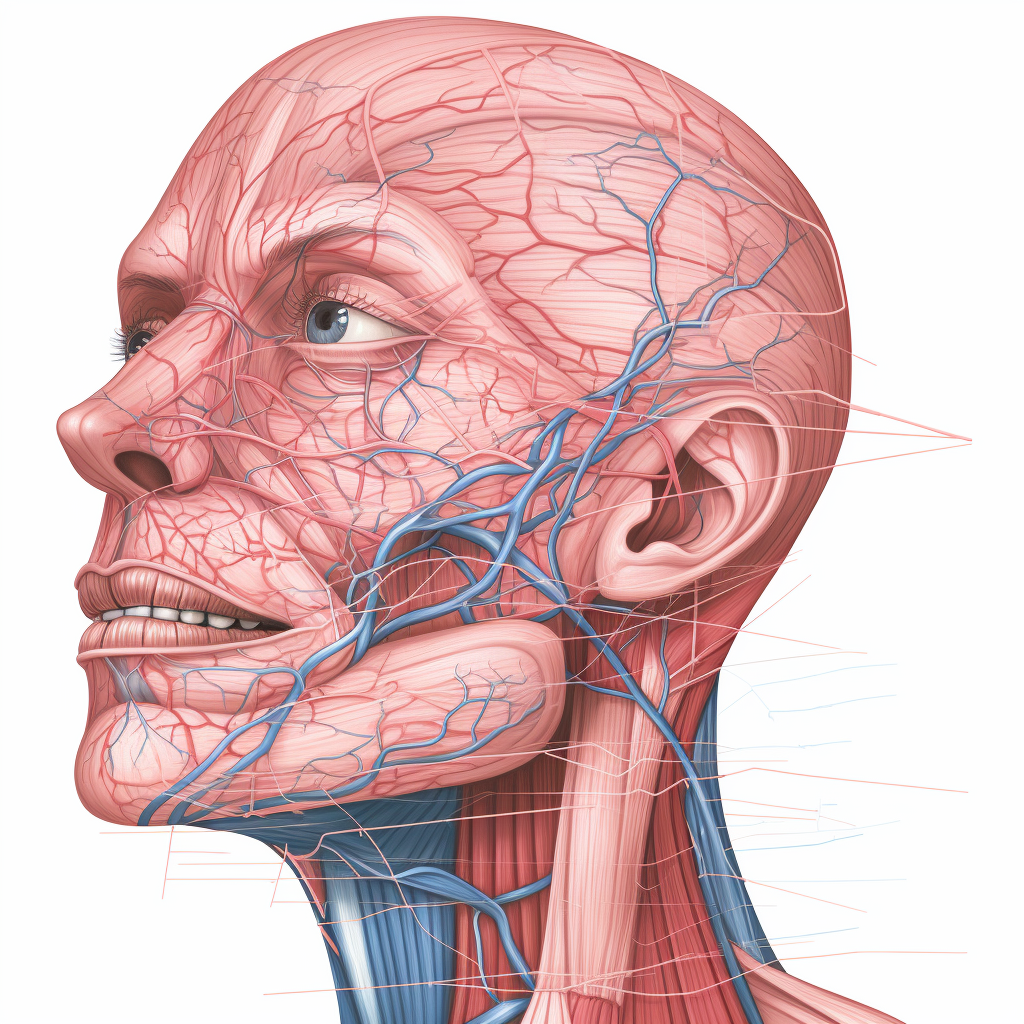
Vena facialis terimi “yüz damarı” anlamına gelen Latince bir kelimedir. “Vena” kelimesi Latince “ven” kelimesinden, “facialis” kelimesi ise Latince “yüze ait” anlamına gelen “facialis” kelimesinden gelmektedir. “Vena facialis” teriminin kayıtlara geçen ilk kullanımı 16. yüzyılda olmuştur. Ön yüz toplardamarı olarak da bilinen yüz toplardamarı (vena facialis), yüzün ön kısmını drene eden bir toplardamardır. Konumu ve … Devamını oku
