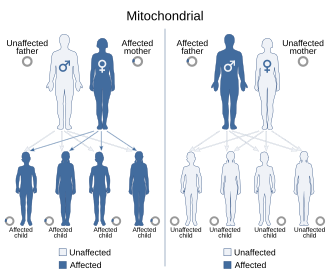
- Döllenme sırasında, sperm sadece Dna’sını yumurtaya verir ve döllenme boyunca yumurtanın organelleri (mitokondri v.s.) kullanılır. Eğer anne mitokondriyal kalıtım hastalığı taşıyorsa, bu hastalığı çocuklarına aktarır. Buna maternal yada mitokondriyal kalıtım denir.
Genetik
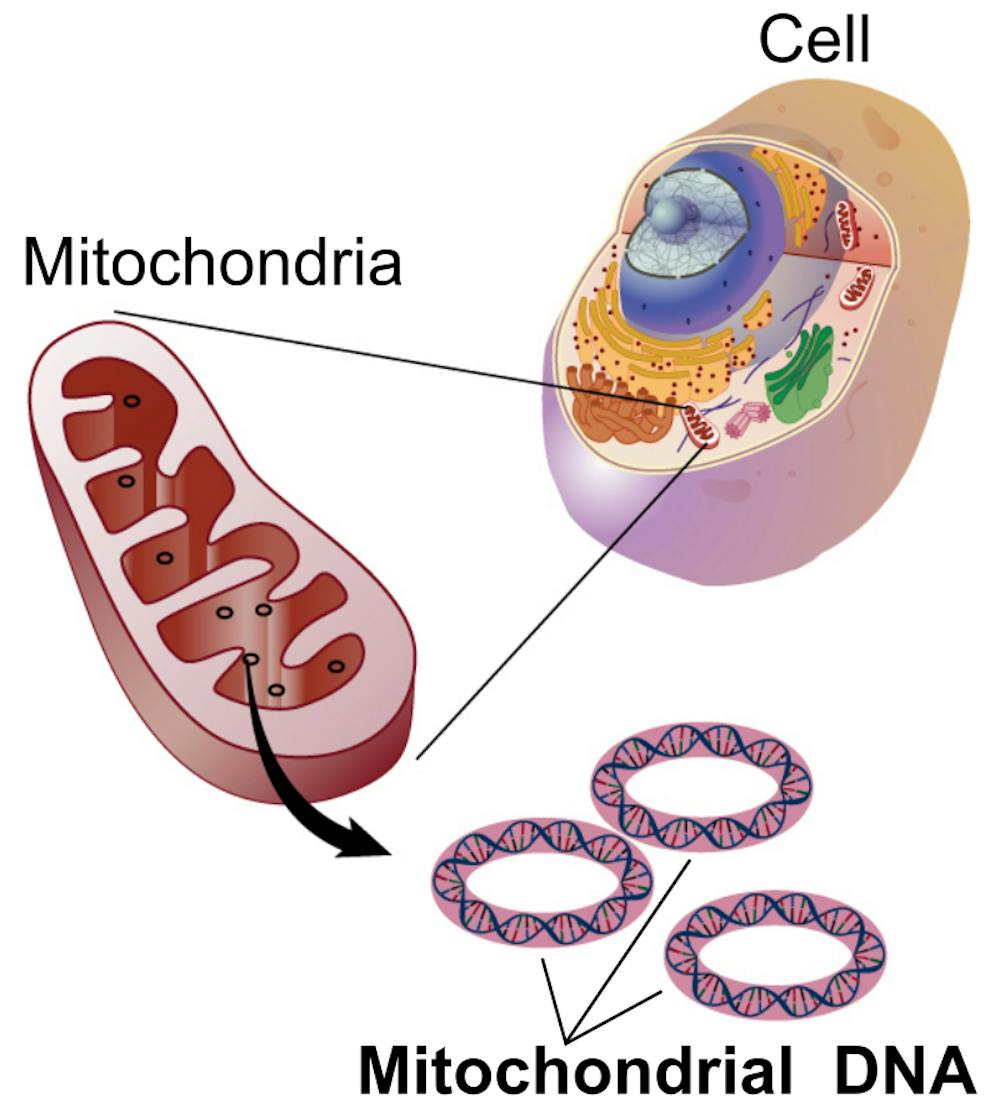
- Kodlayan DNA, ökaryotik hücrelerde sadece nükleer genomda değil, aynı zamanda mitokondride bulunur. Mitokondriyal genomdan veya mitokondriyal DNA’dan (mtDNA) terimleri ile ifade edilir.
- Mitokondri başına 2-10 kopya halinde yaklaşık 16.5 kb (kilobaz) boyutunda halka şeklinde bir çift iplikçik olarak mevcuttur.
- Ökaryotik hücrelerin çoğu, yüzlerce mitokondriye dağılmış 1.000 mtDNA’dan fazla molekül içerir.
- Olgun yumurta hücresi genellikle 100.000’den fazla mtDNA kopyası içerir ve bu da olgun bir yumurta hücresinin toplam DNA’sının üçte birini oluşturur.
- MtDNA molekülü, 13’ü solunum zincirindeki proteinleri kodlayan (oksidatif fosforilasyon) toplam 37 gen içerir. Geri kalan 24 gen, 13 mitokondriyal olarak kodlanmış polipeptidin protein biyosentezi için gerekli olan 22 tRNA ve 2 rRNA molekülünü kodlar.
- Diğer tüm mitokondriyal proteinler ise nükleer olarak kodlanır ve sitosolik sentezden sonra mitokondriye taşınır. Solunum zincirindeki 74 proteinin çoğunluğu (87’nin 74’ü) nükleer genom tarafından kodlanır. Çoğu mitokondriyal hastalığın nükleer genomdaki mutasyonlara dayandırılmasının nedeni de budur.
- Histon (naked Dna=çıplak Dna) ve intron bulunmaz (polycistronik).
Hidroksi radikallerin salınımı
- Mitokondriyal genom, mitokondriyal proteini kodlar. 500 Polimorfizm, 50 olası patojen mutasyon, 55 bilinen patojen mutasyon, Poliploidi (homo- vs. heteroplasmik), değişken Penetranz ve buna göre değişen klinik.
- Eğer mitokondrinin bir kısmında sorun varsa Heteroplazmi, diğer yandan, sadece normal veya mutasyona uğramış mtDNA ile mitokondri içeren hücreler varsa, bir homoplazmiden bahsedilir.
- Mitokondriyal mutasyon solunum zincirinin alt elemanlarını veya direk mitokondriyal sentezlenmiş tüm proteinleri(t-RNA veya r-RNA) etkileyebilir.

Mitokondriyal genomun bir başka özelliği, çoğalmadan sonra yeni oluşturulmuş mitokondriye rastgele dağıtılması ve bunların hücre bölünmesi sırasında ortaya çıkan kızı hücrelere aktarılmasıdır. Nükleer genom için olduğu gibi, sıkı kontrol edilen bir dağıtım mekanizması mtDNA için mevcut değildir. Çoğalmaları da hücre döngüsünden bağımsızdır.

Mitokondriyal Mutasyonlar: İnsan Genetik Bozuklukları
- Mitokondrinin genetiği hakkındaki bilgimiz şimdi büyük ölçüde genişledi. İnsan mitokondrilerinde bulunan DNA tamamen dizilenmiştir ve 16.569 baz çifti içerir. Mitokondriyal gen ürünleri tanımlanmıştır ve aşağıdakileri içerir:
- Aerobik hücresel solunum için gerekli 13 protein
- Çeviri için gerekli 22 transfer RNA’sı (tRNA’lar)
- Çeviri için gerekli 2 ribozomal RNA (rRNA’lar)
- Bir hücrenin enerji kaynağı büyük ölçüde aerobik hücresel solunuma bağlı olduğundan, herhangi bir mitokondriyal genin mutasyonla bozulması, bu organizma üzerinde potansiyel olarak ciddi bir etkiye sahip olabilir.
- Aslında mtDNA, iki olası nedenden dolayı mutasyonlara karşı özellikle savunmasızdır.
- İlk olarak, mtDNA hasarını onarma yeteneği nükleer DNA’nınkine eşdeğer görünmüyor.
- İkincisi, bu kadar sınırlı bir alanda biriken hücre solunumu ile üretilen yüksek derecede mutajenik serbest radikallerin konsantrasyonu, muhtemelen mtDNA’daki mutasyon oranını yükseltir.
- Neyse ki, bir zigot yumurta yoluyla çok sayıda organel alır, bu nedenle sadece bir organel veya birkaçında bir mutasyon varsa (heteroplazminin bir örneği), etki, mutasyona sahip olmayan ve normal şekilde işlev gören birçok mitokondri tarafından büyük ölçüde seyreltilir.
- İlk organel popülasyonunda zararlı bir mutasyon ortaya çıkarsa veya mevcutsa, yetişkinler hem normal hem de anormal organellerin değişken bir karışımına sahip hücrelere sahip olacaktır. Genetik açıdan bakıldığında, bu heteroplazi durumu analizi oldukça zorlaştırır. İnsanlardaki birçok bozukluğun mitokondriyal genlerdeki mutasyonlardan kaynaklandığı bilinmektedir.
- Örneğin, miyoklonik epilepsi ve düzensiz kırmızı lif hastalığı (MERRF), anneden bulaşma ile tutarlı bir kalıtım modeli gösterir. Bu bozukluğu yalnızca etkilenen annelerin çocukları miras alırken, etkilenen babaların yavruları normaldir.
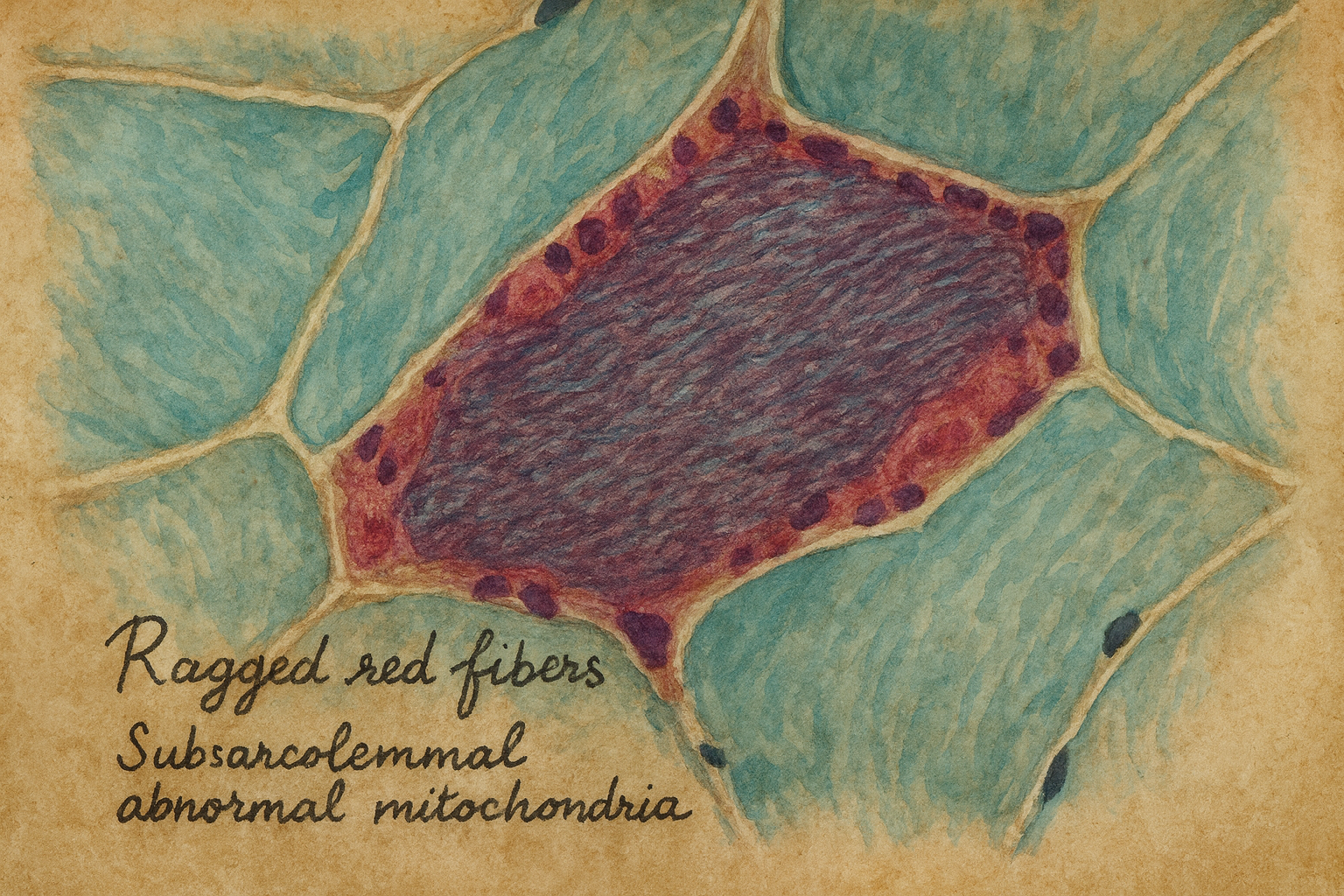
- Bu nadir bozukluğu olan kişilerde ataksi (kas koordinasyon eksikliği), sağırlık, demans ve epileptik nöbetler görülür. Hastalığın adı, anormal mitokondrinin çoğalmasından kaynaklanan lekeli kırmızı lekeler sergileyen “düzensiz kırmızı” iskelet kası liflerinin varlığından kaynaklanmaktadır.
- Bu bozuklukta enerji ihtiyacı yüksek olan beyin fonksiyonu da etkilenerek yukarıda tarif edilen nörolojik semptomlara yol açar.
- MERRF’li hastalardan alınan mtDNA analizi, bir transfer RNA’sını kodlayan 22 mitokondriyal genden birinde bir mutasyon ortaya çıkardı. Spesifik olarak, tRNALys’i kodlayan gen (translasyon sırasında lizin sağlayan tRNA) değiştirilmiş bir DNA sekansı içerir. Bu genetik değişiklik, organeldeki çeviri kapasitesine müdahale eder ve bu da bozukluğun çeşitli tezahürlerine yol açar. Etkilenen bireylerin hücreleri, normal ve anormal mitokondri karışımını içeren heteroplazma adı verilen durumu sergiler. Farklı hastalar, ikisinin farklı oranlarını içerir ve hatta aynı hastadan farklı hücreler, çeşitli düzeylerde anormal mitokondri sergiler.
- Örneğin, miyoklonik epilepsi ve düzensiz kırmızı lif hastalığı (MERRF), anneden bulaşma ile tutarlı bir kalıtım modeli gösterir. Bu bozukluğu yalnızca etkilenen annelerin çocukları miras alırken, etkilenen babaların yavruları normaldir.
- Heteroplazi olmasaydı, mutasyon büyük olasılıkla ölümcül olurdu, mitokondriyal fonksiyonun temel doğasına ve organel içinde mtDNA tarafından kodlanan genlere bağlılığına tanıklık ederdi.

Klinik
- Mitokondriyal mutasyonlar semptomatik hastalarda genellikle sadece bir hücrenin mitokondrilerinin bir kısmında bulunur.
- Bir mitokondriyal mutasyonun aslında bir fenotipik etkiye sahip olup olmadığı, hücrelerdeki normal ve mutasyona uğramış mtDNA arasındaki orana bağlıdır. Eksik penetrasyon, değişken ekspresyon ve pleiotropi bu nedenle tüm mitokondriyal kalıtsal hastalıkların tipik özellikleridir. Normal ve mutasyona uğramış mtDNA kopyaları arasındaki oran sadece farklı organlar arasında değil, aynı zamanda birkaç hücre bölünmesi boyunca da değişebilir. Bu nedenle mitokondriyal kalıtsal hastalıkları olan hastaların sıklıkla değişen semptomları tanımlaması tipiktir.
- Eğer solunum zincirinin bir elemanında sorun çıkarsa, hücrenin enerji temini değişir ve yüksek enerji ihtiyacı açığa çıkar. Etkilenenler artık ışığa maruz kalsalar bile oksidatif fosforilasyon ile enerji gereksinimlerini karşılayamazlar. Sonuç olarak, giderek daha fazla laktik asidoz alırlar. Ek olarak, iskelet kaslarında mikroskopik sözde düzensiz kırmızı lifler – subarkomal mitokondri birikimleri bulunur.
- Tipik Mitokondriyal kalıtım hastalıkları:
- Ensefalopati,
- Miyopati,
- Kardiyomiyopati,
- Ataksi,
- Retinal dejenerasyon ve dış göz kaslarının felci,
- Hafif olarak vücut zorlandıktan sonra Laktat asidozu ve iskelet kası ragged red fibers =bozuk mitokondrilerin subsarkolemmalda toplanması.
- MELAS-Sendromu; Kısaltma olarak kullanıldığında, miyopati, ensefalopati, laktik asidoz ve inme benzeri atakları ifade eder. Tipik olarak mitokondriyal tRNALeu genindeki 3243A G mutasyonundan kaynaklanır.
- MERRF-Sendromu; Düzensiz Kırmızı Lifli Miyoklonik Epilepsi anlamına gelir. Tipik olarak mitokondriyal tRNALys genindeki 8344G A mutasyonundan kaynaklanır.
- NARP sendromu: Burada harfler şu anlama gelir: nöropati, ataksi ve retinitis pigmentoza. NARP sendromuna çoğunlukla ATPase genindeki 8993T G veya TC mutasyonu neden olur.
- CPEO
- Kearns-Sayre-Syndromu
- Pearson-Syndromu
- LHON (Karaciğer Kalıtsal Optik Nöropati) mitokondriyal kalıtsal hastalıklar arasında bir istisnadır. Bu multi sistemik etkili bir hastalık değildir. Sadece optik sinir etkilenir. Bunun ortak nedeni, mitokondriyal DNA’daki 11778G A mutasyonudur.
Mitokondrilerdeki mutasyonların her zaman hücresel enerji metabolizması üzerinde etkisi vardır, bu nedenle kalp, karaciğer, böbrek, kaslar ve SSS gibi yüksek enerji devirine sahip organlar özellikle etkilenir. Mutasyonların oranı birkaç hücre bölünmesi boyunca da değiştiğinden, mitokondriyal kalıtsal hastalıkların semptomları da sıklıkla değişir.
Mitokondriyal Hipotez, yaşlanma süreci, Parkinson, M. Alzheimer, toksik „serbest“ Radikale (ROS) Mutagenez, Tumorigenez konuları ile ilişkileri hala tartışılmaktadır.
Tarih
- Mitokondriyal DNA 1960’larda Margit M. K. Nass ve Sylvan Nass tarafından elektron mikroskopisi ile mitokondri içindeki DNaz’a duyarlı iplik olarak keşfedildi.
- Ellen Haslbrunner, Hans Tuppy ve Gottfried Schatz tarafından biyokimyasal testlerle yüksek saflaştırılmış mitokondriyal fraksiyonlar üzerinde keşfedildi




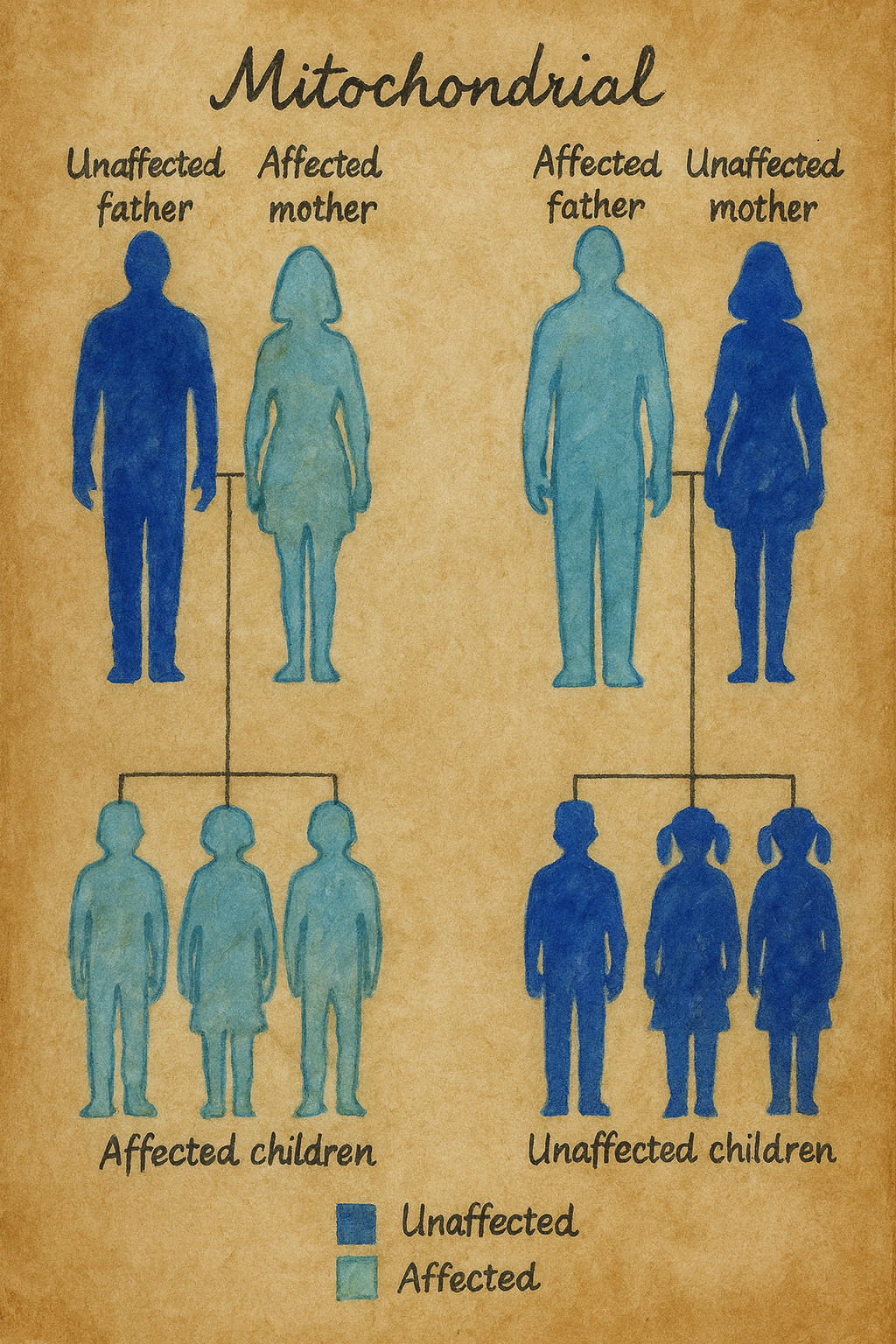
 gereken cinsiyette görülmüşse, sıkça tekrar etmesini ifade eder.
gereken cinsiyette görülmüşse, sıkça tekrar etmesini ifade eder.
Yorum yazabilmek için oturum açmalısınız.