İçindekiler
Tanım ve Temel Kavramlar
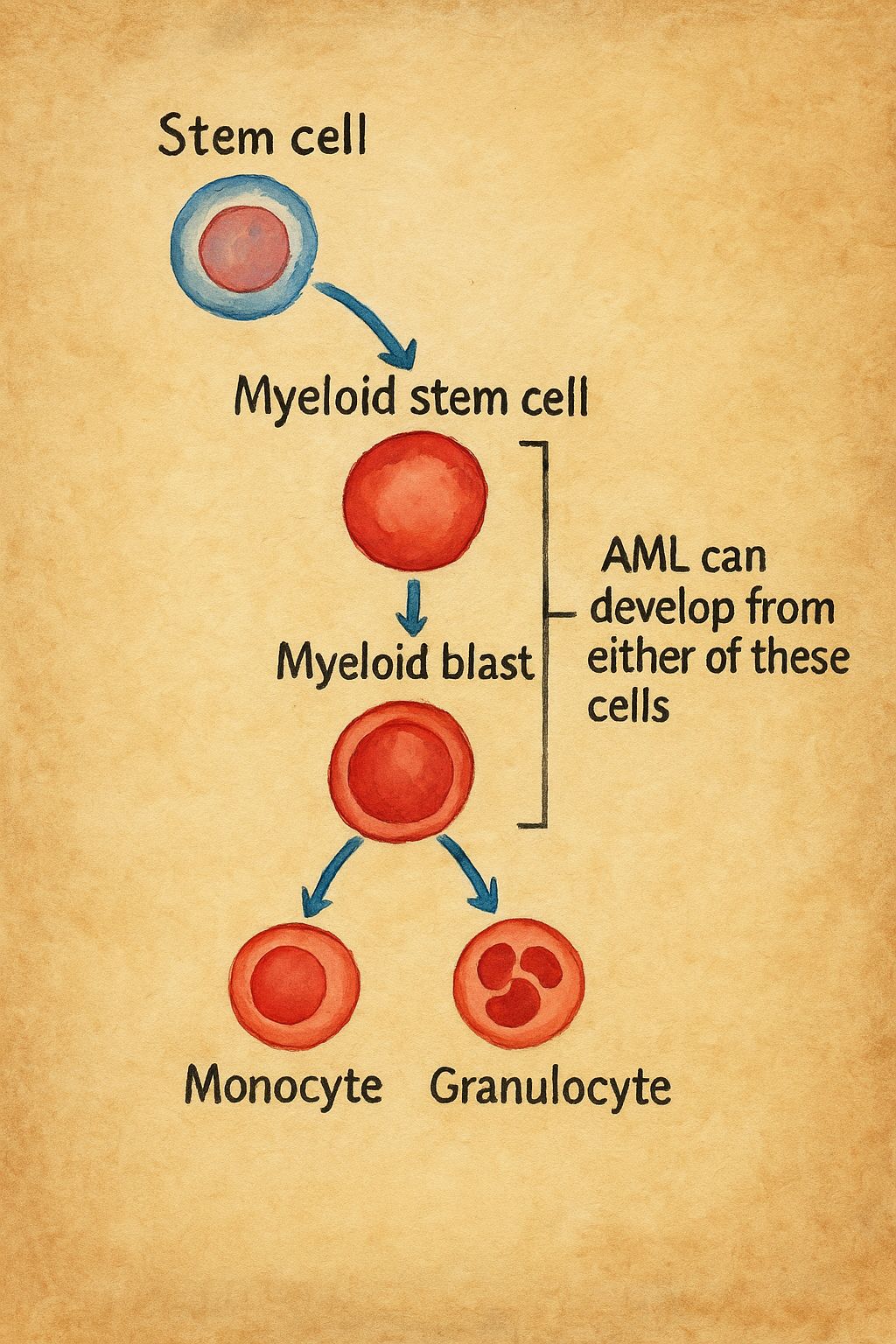
Akut miyeloid lösemi (AML), miyeloid soy hattına ait hematopoetik öncül hücrelerin (miyeloblastlar ve daha ileri promiyelosit–miyelosit evreleri) kontrolsüz proliferasyonu ve farklılaşma blokajı ile karakterize, hızlı seyirli bir kan ve kemik iliği neoplazisidir. Klinik olarak kemik iliği yetmezliği bulguları (anemi, trombositopeni, nötropeni) ve/veya hiperleukositozla ilişkili komplikasyonlarla belirti verir.
Klasik tanı eşiği, kemik iliğinde veya periferik kanda ≥%20 blast varlığıdır; bununla birlikte güncel sınıflandırmalar, belirli genomik anormalliklerin (ör. RUNX1::RUNX1T1 [t(8;21)], CBFB::MYH11 [inv(16)/t(16;16)], PML::RARA [t(15;17)], bazı KMT2A yeniden düzenlenmeleri, inv(3)/t(3;3), t(6;9) gibi) varlığında blast yüzdesi <%20 olsa dahi AML teşhisine izin verebilmektedir.
Terimler ve Kısaltmalar
- MRD: Ölçülebilir artık hastalık
- HMA: Hipometilleyici ajan (azasitidin, desitabin)
- HiDAC: Yüksek doz sitarabin
- allo-HHN: Allojenik hematopoetik hücre nakli
- APL: Akut promiyelositik lösemi
- DİK: Damar içi yaygın pıhtılaşma
Epidemiyoloji
AML erişkin akut lösemilerin ~%80’ini oluşturur; medyan tanı yaşı 65–70 yıl civarındadır. Toplam yıllık insidans gelişmiş ülkelerde ~3–5/100.000’dir; yaşla birlikte belirgin biçimde yükselir ve ileri yaş gruplarında 20–30/100.000 düzeylerine ulaşabilir. Erkeklerde hafif artmış sıklık bildirilir. Tütün kullanımı, benzen maruziyeti ve iyonize radyasyon gibi çevresel faktörler; terapiye bağlı olgular (alkilleyiciler, topoizomeraz II inhibitörleri sonrası) ve öncül klonal bozukluklar (MDS, MDS/MPN) risk artışıyla ilişkilidir.
Etiyopatogenez ve Moleküler Temeller
AML, çok basamaklı bir klonal evrim sürecinin sonucudur:
- Sürücü mutasyonlar ve sitogenetik değişiklikler işlevsel olarak birkaç eksende toplanır:
- Proliferasyon sinyallemesi (Sınıf I): FLT3 (ITD/TKD), KIT, NRAS/KRAS.
- Transkripsiyon ve farklılaşma (Sınıf II): NPM1, RUNX1, CEBPA (özellikle bZIP bölge mutasyonları).
- Epigenetik düzenleyiciler: DNMT3A, TET2, IDH1/2, ASXL1, EZH2.
- Spliceosom: SRSF2, U2AF1, SF3B1, ZRSR2.
- Kohezin kompleksi: STAG2, SMC1A/SMC3.
- Tümör baskılayıcılar: TP53 (çoğunlukla kompleks/monozomal karyotiple birlikte).
- Klonal hematopoez (CHIP/CCUS) zemininde, özellikle DNMT3A, TET2, ASXL1 mutasyonları taşıyan yaşlı bireylerde, ek sürücü olayların birikimiyle ikincil/yaşa bağlı AML gelişebilir.
- Terapiye bağlı AML (t-AML) genellikle kompleks karyotip, TP53 mutasyonu veya KMT2A yeniden düzenlenmesi taşır ve prognozu olumsuzdur.
- APL (akut promiyelositik lösemi), PML::RARA füzyonuna bağlı özgün bir alt tip olup yaygın DİK ile seyredebilir; patofizyolojide retinoik asit reseptör sinyallemesinin baskılanması ve farklılaşma blokajı söz konusudur.
Morfoloji ve İmmünofenotip
- Periferik yayma/kök iliği: Büyük çekirdekli, ince kromatinli, nükleoluslu blastlar; sıklıkla Auer çubukları (özgül granül lizozomal kristaloidlerinin iğne benzeri birikimi).
- Sitokimyasal boyamalar: Miyeloperoksidaz (MPO) ve Sudan Black B sıklıkla pozitiftir (özellikle granülositik soy).
- Akım sitometrisi: CD13, CD33, CD117, MPO, HLA-DR (APL’de sıklıkla negatif), CD34 (değişken), monositik alt tiplerde CD14/CD64; CD33 hedefliliği tedavi seçimini etkileyebilir (gemtuzumab ozogamisin).
- APL’de “faggot cells” olarak betimlenen çok sayıda Auer çubuklu hipergranüler promiyelositler karakteristiktir.
Klinik Özellikler
- Kemik iliği yetmezliği:
- Anemi → solukluk, efor dispnesi, çarpıntı.
- Trombositopeni → peteşi, purpura, burun/diş eti kanaması, menstrüel artış.
- Nötropeni/disfonksiyon → ateş, oral kandidiyazis, bakteriyel ve invaziv fungal enfeksiyonlar.
- Hiperleukositoz/leukostasis (özellikle monositik varyantlarda): dispne, hipoksemi, nörolojik semptomlar; acil sitoredüksiyon gerekebilir.
- Organ infiltrasyonu: Splenomegali, hepatomegali; gingival hipertrofi (M4/M5), kutanöz lösemi, miyeloid sarkom (klorom).
- APL’de koagülopati/DİK: Fibrinojen düşüklüğü, D-dimer yüksekliği, yaygın kanamalar.
Ayırıcı Tanı
- MDS-EB (özellikle %10–19 blast aralığı), ALL, CML blast krizi, reaktif blastik cevaplar (nadiren). İmmünofenotip, sitogenetik ve moleküler analiz belirleyicidir.
Tanı Yaklaşımı
- Tam kan sayımı ve yayma: Anemi, trombositopeni, lökositlarda düzensizlik; hiatus leucaemicus gözlenebilir.
- Koagülasyon testleri: APL şüphesinde fibrinojen, PT/aPTT, D-dimer acil değerlendirilir.
- Biyokimya: LDH, ürik asit (tümör lizisi riski), böbrek/karaciğer fonksiyonları.
- Kemik iliği aspirasyon/biopsi: Morfoloji, sitokimya, akım sitometrisi, karyotip/FISH, NGS panel (en az NPM1, FLT3, CEBPA, TP53, IDH1/2, RUNX1, ASXL1, DNMT3A vb.).
- MRD değerlendirimi: Çok parametreli akım ve/veya moleküler (ör. NPM1 kopya sayımı, füzyon transkriptleri) ile tedavi yanıtının derinliği izlenir.
- HLA tiplemesi: Allojenik nakil adaylarında tanı anında planlanmalıdır.
Sınıflandırma
1) FAB (Geleneksel Morfolojik)
- M0 minimal farklılaşmış, M1 olgunlaşmasız, M2 olgunlaşan miyeloblastik;
- M3 APL; M4 miyelomonositik (± anormal eozinofili: M4Eo);
- M5a/b monoblastik/monositik; M6 eritroid baskın; M7 megakaryoblastik.
Güncel uygulamada prognostik ve terapötik kararlar moleküler-sitogenetik temelde verildiğinden FAB ikincil önemdedir.
2) Güncel Moleküler Tabanlı (WHO 2022 ve ICC 2022 ana hatlar)
- AML, tanımlayıcı genetik anormalliklerle (RUNX1::RUNX1T1, CBFB::MYH11, PML::RARA, belirli KMT2A/MECOM/DEK::NUP214 vb.).
- AML, NPM1-mutasyonlu ve AML, bialelik CEBPA mutasyonlu (özellikle bZIP bölge).
- AML, miyelodisplazi ile ilişkili (AML-MR): belirli MR gen mutasyonları, özgün sitogenetik diziler ve/veya MDS öyküsü ile.
- Terapiyle ilişkili miyeloid neoplaziler (t-MN): t-AML/t-MDS.
- AML, NOS: özgül genetik belirteç yokluğunda morfolojiye göre.
Not: Eşik blast yüzdesi ve “genetikle tanımlı” AML ölçütleri WHO ve ICC çerçevelerinde nüanslar içerebilir; pratikte kurumunuzun kabul ettiği kılavuza uyum esastır.
Risk (Prognostik) Sınıflama
ELN temelli risk katmanlaması, sitogenetik ve moleküler profilin birleşimiyle olumlu / ara / olumsuz gruplar tanımlar.
- Olumlu: RUNX1::RUNX1T1, CBFB::MYH11, NPM1 mut (advers karyotip eşlik etmiyorsa), bialelik CEBPA mut.
- Ara: Özgül olumlu/olumsuz kriterlere girmeyenler; FLT3 değişimleri risk-modifiye edici olabilir.
- Olumsuz: TP53 mut/abn(17p), kompleks/monozomal karyotip, inv(3)/t(3;3), t(6;9), belirli KMT2A R’leri, -5/del(5q), -7/del(7q) ve AML-MR ile ilişkili imzalar.
MRD durumu, tedavi/aktarım kararlarını güçlü biçimde etkiler ve risk tahminini dinamik olarak günceller.
Tedavi
Acil Destek ve Özel Durumlar
- APL şüphesi: Koagülopatiyi sınırlamak için ATRA gecikmeksizin başlanır; yüksek riskte arsenik trioksit ve/veya antrasiklin eklenir. Diferansiyasyon sendromu olasılığına karşı steroid profilaksisi/tedavisi planlanır.
- Hiperleukositoz/leukostasis: Hidroksiüre ile sitoredüksiyon, seçilmiş olgularda lökaferez; TLS profilaksisi (hidrasyon, allopurinol/rasburikaz).
- Enfeksiyon profilaksisi: Antibakteriyel+antifungal stratejiler, febril nötropeni protokolleri, transfüzyon eşikleri (Hb, trombosit).
İndüksiyon (Küratif Niyet)
- “7+3”: Sitarabin (7 gün) + antrasiklin (daunorubisin/idarubisin, 3 gün) standart yaklaşımdır.
- FLT3-mutasyonlu olgularda midostaurin indüksiyon/konsolidasyona eklenebilir.
- CD33-pozitif, uygun karyotipte (özellikle çekirdek bağlayıcı faktör AML) gemtuzumab ozogamisin eklenmesi nüks riskini azaltabilir.
- t-AML/MDS-ilişkili AML’de CPX-351 (daunorubisin+sitarabin liposomal) üstünlük gösterebilir.
Konsolidasyon
- Olumlu riskli genç hastalarda yüksek doz sitarabin (HiDAC) sekansları sıktır.
- Ara/olumsuz risk veya kalıcı MRD varlığında, ilk tam remisyonda allojenik hematopoetik hücre nakli (allo-HHN) kuvvetle düşünülür.
Yaşlı / Kırılgan Hastalar
- Venetoklaks + hipometilleyici ajan (azasitidin veya desitabin) ya da venetoklaks + düşük doz sitarabin, yüksek yanıt oranları ve MRD negatiflikleri ile standartlaşmıştır.
- Seçilmiş olgularda glasdegib + LDAC gibi kombinasyonlar da kullanılabilir.
Hedefe Yönelik ve Kurtarma Tedavileri
- FLT3: Gilteritinib (özellikle R/R AML’de); FLT3-pozitif ilk hatta bazı bölgelerde midostaurin/quizartinib seçenekleri.
- IDH1/IDH2: Ivosidenib / enasidenib (monoterapi veya kombinasyon).
- CD33: Gemtuzumab ozogamisin (uygun profilde).
- Menin inhibitörleri (KMT2A R / NPM1 mut AML) ve yeni ajanlar (anti-CD47, TIM-3 vb.) klinik geliştirme aşamasındadır.
- Nüks: Moleküler hedefe göre ilaçlar, yeniden indüksiyon, köprüleme ve allo-HHN değerlendirmesi.
İzlem ve MRD
- MRD değerlendirmesi indüksiyon sonrası, her konsolidasyon sonrası ve idame/izlem döneminde belirli aralıklarla yapılır. NPM1 transkript izlemi, RUNX1::RUNX1T1/CBFB::MYH11 kopya izlemi ve akım MRD en yaygın yaklaşımlardır. MRD ** negatifliği**, nüks riskini düşürür ve nakil kararlarını şekillendirir.
Komplikasyonlar
- Enfeksiyonlar (bakteriyel, invaziv küf): mortalitenin temel belirleyeni; preemptif/ampirik yaklaşımlar kritik.
- Kanama/DİK (özellikle APL): fibrinojen destekleri ve hemostaz yönetimi.
- Tümör lizis sendromu: elektrolit bozuklukları, AKI; profilaksi ve yakın izlem şarttır.
- Kardiyotoksisite (antrasiklinler), mukozit, nörotoksisite (yüksek doz Ara-C), infertilite.
- Nakil ilişkili: GVHH, enfeksiyonlar, uzun dönem sekeller.
Prognoz
- Tam remisyon (CR): yoğun indüksiyon alan <60 yaş erişkinlerde sıklıkla %60–80; ileri yaşta daha düşüktür.
- Uzun dönem sağkalım: risk grubuna, MRD durumuna ve nakil stratejisine bağlı olarak genç erişkinlerde ~%40–60, ileri yaşta ~%15–25 aralığındadır.
- APL güncel tedavilerle > %80–90 kalıcı kür oranlarına ulaşır.
- Olumsuz belirteçler: ileri yaş, yüksek başlangıç lökositi/hiperleukositoz, kalıcı MRD, TP53 mutasyonu/kompleks karyotip, terapiyle ilişkili veya MDS-ilişkili genetik imza.
Özel Durumlar
- CNS tutulumu: Tanıda nadirdir; monositik alt tip ve çok yüksek lökositte risk artar. Klinik/BTK bulgularında lomber ponksiyon ve intratekal tedavi gerekebilir.
- Gebelik: Trimester, anne/fetüs riski ve alt tipe göre multidisipliner planlama; ATRA/ATO sınırlamaları özellikle önemlidir.
- Miyeloid sarkom: İzole “ekstramedüller” tutulum dahi sistemik AML gibi tedavi edilir.
- Destek tedavileri: Antimikrobiyal profilaksi, büyüme faktörleri seçilmiş durumlarda, transfüzyon eşikleri ve yaşam tarzı/rehabilitasyon bileşenleri sonuçları etkiler.
Pratik Tanı–Tedavi Akış Özeti
- Şüphe (sitopeniler ± blastlar ± kanama/enfeksiyon) → acil kan yayması ve koagülasyon; APL düşünülen her hastaya ATRA başla.
- Kemik iliği + akım + genetik/NGS → AML tanısı ve risk sınıflaması.
- Genç/fit: 7+3 ± hedefe yönelik ajanlar → MRD yönelimli konsolidasyon (HiDAC veya allo-HHN).
- Yaşlı/kırılgan: Venetoklaks + HMA/LDAC → yanıt derinliğine göre sürdürme/nakil değerlendirmesi.
- Nüks/R/R: Moleküler hedefe yönelik ilaçlar ± yeniden indüksiyon ± allo-HHN.
- Boyunca destek tedavi ve MRD izlemi.
Keşif
Akut miyeloid löseminin (AML) hikâyesi, kanın mikroskop altında ilk kez “olağandışı beyazlıkta” göründüğü kayıtlardan, tek hücreli dizilemenin, minimal artık hastalık (MRD) izleminin ve yeni hedefe yönelik-epigenetik ilaçların şekillendirdiği günümüz kliniklerine uzanan, iki asırlık bir bilim ve tıp serüvenidir.
İlk işaretler: “Beyaz kan”ın doğuşu (1811–1847)
- yüzyıl başında tekil olgu bildirimlerinde hekimler, dalak büyümesi ve sıra dışı “süt renginde” kanla seyreden vakalar tanımladı; Alfred Velpeau ve Alfred Donné’nin mikroskopi merakı, kandaki “renksiz tanecikler”e (lökositler) dikkat çekilmesine zemin hazırladı. 1845’te Edinburgh’da John Hughes Bennett, kanında olağandışı lökosit artışı olan bir hastayı ayrıntılı biçimde yayınlayıp hastalığı “leukocythemia” diye adlandırdı; aynı yıl Berlin’de Rudolf Virchow benzer gözlemler yaptı ve 1847’de “Leukämie” terimini önererek hastalığı bir kan hastalığı olarak kavramsallaştırdı.
Kemik iliğine iniş: Hastalığın kaynağı beliriyor (1868–1880)
1868’den itibaren Königsberg’li patolog Ernst Christian Neumann, postembriyonik dönemde hem eritropoez hem lökopoezin kemik iliğinde gerçekleştiğini gösterdi; kemik iliğini kanın “fidanlığı” olarak tanımladı ve “miyelojenöz lösemi” kavramını yerleştirdi. Aynı dönemde Paul Ehrlich’in anilin boyaları kullanarak geliştirdiği ayırıcı boyama yöntemleri, granülosit serilerinin ve blast morfolojisinin ayrımını mümkün kıldı; hematolojide hücre temelli modern sınıflamanın kapısı açıldı.
Mikroskobun imzası: Auer çubukları ve morfoloji (1905–1930’lar)
1905’te Thomas McCrae ve 1906’da John Auer, bugün adını Auer’den alan, miyeloid patlamaların sitoplazmasındaki iğnemsi kristalleri tanımladı; bu inklüzyonlar özellikle AML ve akut promiyelositik lösemide (APL) miyeloid kökenin patognomonik ipuçlarından biri olarak kabul gördü. 20. yüzyılın ilk yarısında, morfoloji ve sitokimya AML tanısının temel omurgasını oluşturdu.
Kemoterapi çağının şafağı: Ara-C ve antrasiklinler (1950’ler–1970’ler)
İkinci Dünya Savaşı sonrasında antimetabolitler ve antibiyotik kökenli antitümör ajanlar klinik sahneye çıktı. Deniz süngerlerinden ilhamla sentezlenen sitarabin (ara-C) 1959’da sentezlendi ve 1969’da onay aldı; İtalya ve Fransa’daki ekiplerin toprak aktinobakterilerinden izole ettiği daunorubisin 1960’larda lösemi tedavisinde hızla benimsendi. 1973’te Yates ve çalışma arkadaşlarının verileri, 7 gün kesintisiz sitarabin + ilk 3 gün antrasiklin (“7+3”) kombinasyonunu AML indüksiyonunun altın standardı olarak biçimlendirdi—bu şablon, yarım yüzyıldır sayısız varyasyonuyla yaşıyor.
Sınıflandırma devrimi I: FAB ve morfolojik dil (1976–1985)
Fransız-Amerikan-İngiliz (FAB) grubu, AML’yi M0–M7 arasında morfoloji ve sitokimya temelinde tanımlayan bir şema yayımladı. Klinik iletişimi standardize eden bu dil, güncel moleküler sınıflamalarla bile gündelik pratikte referans olmaya devam ediyor.
Kromozomal imzalar: Çekirdek bağlayıcı faktörler ve APL (1970’ler–1990’lar)
1970’lerde sitogenetiğin hassaslaşmasıyla AML’nin “tekrarlayan” anomalileri belirmeye başladı: t(8;21) RUNX1-RUNX1T1 ve inv(16)/t(16;16) CBFB-MYH11, “çekirdek bağlayıcı faktör (CBF)-AML” başlığı altında iyi riskli bir biyoloji tanımladı. APL’de t(15;17) anomalisi 1977’de netleşti; 1991’de PML-RARA füzyonunun keşfi, farklılaşma tedavisinin biyolojik temelini kurdu.
Bir hastalığın kaderi değişiyor: ATRA ve arsenik (1985–2010’lar)
1980’lerin ortasında Şanghay’da Zhen-Yi Wang ve ekibi, APL’de all-trans retinoik asidin (ATRA) dramatik farklılaştırıcı etkilerini bildirdi; 1990’larda arsenik trioksit (ATO) ile birleşen bu yaklaşım, APL’yi “yüksek ölümcül” bir tablodan “yüksek oranda kür” alınabilen bir akut lösemiye dönüştürdü. 2013 APL0406 çalışması, düşük-orta riskli APL’de kemoterapisiz ATRA+ATO rejiminin etkinliğini randomize olarak göstererek paradigma değişimini taçlandırdı.
Sınıflandırma devrimi II: WHO ve ELN çağında genomik AML (2001–2022)
2000’lerden itibaren Dünya Sağlık Örgütü (WHO) sınıflamaları morfolojiye sitogenetik ve moleküler verileri kademeli olarak eklerken, Avrupa Lösemi Ağı (ELN) tedavi ve risk sınıflamasını genetik temelde güncelledi. 2022 baskıları, miyeloid neoplazilerin bilgi çağındaki haritasını belirginleştirerek mutasyon-temelli risk çerçevesini ve yanıt tanımlarını (MRD içeren CR kategorileri dâhil) standardize etti.
Moleküler dönüm noktaları: Sürücü mutasyonlardan hedefe yönelik tedavilere (1996–2025)
1990’ların ortasında FLT3-ITD’nin keşfi; 2000’lerde NPM1, CEBPA; 2009–2011 aralığında IDH1/2, TET2, DNMT3A ve ardından “splicing” ve “cohesin” kompleksi mutasyonları, AML biyolojisini çoklu yollarla şekillendiren bir mutasyon coğrafyası ortaya koydu. Bu harita, günümüz tedavilerini yönlendiren hedeflere dönüştü: FLT3 inhibitörleri (ör. midostaurin, gilteritinib, 2023’te quizartinib), IDH1/2 inhibitörleri (ivosidenib, enasidenib; ardından olutasidenib) ve daha yaşlı/“uygunsuz” hastalarda venetoklaks-hipometilleyici kombinasyonları, standartların parçası hâline geldi. Sekonder/tedavi-ilişkili AML’de lipozomal daunorubisin+sitarabin (CPX-351) yaklaşımı yeni bir kemoterapi paradigması sundu.
Hastalığı “neredeyse görünmez” hâle getirmek: MRD ve ölçülebilir kalıntı (2018–2025)
MRD kavramı, ELN’nin 2021–2022 güncellemeleriyle AML’de teknik-klinik bir standart kazandı: uygun hastalarda NPM1 RT-qPCR ve/veya çok renkli akım-sitometri ile zamanlanmış ölçümler, konsolidasyonun derinliğini ve nüks riskini öngörmede merkezî rol üstlendi; yeni kılavuzlar NGS-temelli MRD’yi de çerçeveledi. Pratikte MRD-negatif tam yanıt, “başarı”yı yalnız morfolojiyle değil biyomoleküler duyarlılıkla tanımlar hâle geldi.
Tek hücre ve mekânsal çok-omik: Klonal mimarinin yüzü (2020’ler)
Tek hücreli DNA/RNA dizileme ve mekânsal transkriptomik, AML’nin kemik iliği ekosisteminde klonların nasıl dalgalandığını, işbirliği yaptığını ve tedaviyle nasıl şekillendiğini gösteren yeni bir “haritalama” sağlıyor. Kompleks karyotipli olgularda aynı hastada birden çok alt klonun eşzamanlı varlığı, pozitif seleksiyon ve karsinogenez yollarının “canlı” izlendiği bir düzeyi mümkün kıldı; MRD çözünürlüğü ve nüks biyolojisinin sezilmesi bu sayede ivme kazandı.
Bağışıklık ve epigenetik cephe: Dalgakıranlar ve sınırlar (2020–2025)
CD47 engelleyicileriyle fagositozun serbest bırakılması ya da TIM-3 hedefli bağışıklık modülasyonu gibi stratejiler dalga dalga klinik denemelere girdi; kimi programlar (ör. magrolimab) güvenlik/etkinlik engellerine takılırken, sınıfın genelinde “kombinasyonla güçlendirme” fikri öne çıktı. Epigenetik hedefler cephesinde “menin inhibitörleri”, KMT2A (MLL) translokasyonlu ve NPM1-mutant lösemilerde oyunun kurallarını değiştirmeye aday oldu; 2024’te revumenib’in FDA onayı, bu hattı somut bir klinik standarda dönüştürdü.
Sürdürme (maintenance) ve nakil ötesi stratejiler: Köprüyü güçlendirmek (2020–2025)
Yoğun tedavi sonrası nüksü geciktirmeye/önlemeye yönelik yaklaşımlar klinik pratiğe yerleşti: 2020’de onaylanan oral azasitidin (CC-486), yoğun tedaviyi tamamlayamayan tam yanıtlı hastalarda genel sağkalımı uzatarak “tabletle sürdürme” devrini açtı. FLT3-ITD pozitif olgularda allojenik nakil sonrası sorafenib sürdürmesi, randomize verilerle nüks ve mortalitede anlamlı azalma sağladı; çok merkezli ve uzun izlemli sonuçlar bu sinyali pekiştirdi.
Bugün ve yarın: Kişiselleştirilmiş, derin ve çevik AML tıbbı
2025 itibarıyla AML, tek bir hastalık değil; farklı sürücülerin, işbirlikçi mutasyonların ve mikroçevre etkileşimlerinin yönettiği bir “miyeloid sendromlar koalisyonu”. Klinik kararlar—indüksiyon yoğunluğu, transplant stratejisi, hedefe yönelik kombinasyon seçimi, MRD eşiği ve izlem sıklığı—artık yaş, performans ve komorbidite kadar genetik-epigenetik haritaya ve biyolojik yanıtın “derinliğine” de dayanıyor. Çekirdek hedeflere (FLT3, IDH1/2) yönelik inhibitörlerin rasyonel bileşimleri; venetoklaks-temelli “yaşlı/uygunsuz” şablonlarında biyobelirteç bazlı rafinman; menin ekseninde farklılaşma-transkripsiyon makinesini “ayarlayan” yeni moleküller; ve tek hücre-mekânsal omiklerin klinik karar destekle entegrasyonu, önümüzdeki on yılın alametifarikası olmaya aday.
İleri Okuma
- Bennett JH. Case of hypertrophy of the spleen and liver in which death took place from suppuration of the blood. Edinburgh Medical and Surgical Journal. 1845;64:413–423.
- Virchow R. Weisses Blut. Froriep’s Notizen aus dem Gebiete der Natur- und Heilkunde. 1845;36:151–156.
- Neumann EC. Das Gesetz der Verteilung der farblosen Blutkörperchen in den Gefäßen und seine Bedeutung für die Blutbildung. Archiv für Heilkunde. 1868;9:107–121.
- Ehrlich P. Beiträge zur Kenntniss der Anilinfärbungen und ihrer Verwendung in der mikroskopischen Technik. Archiv für mikroskopische Anatomie. 1877;13:263–277.
- Auer J. Some hitherto undescribed structures found in the large lymphocytes of a case of acute leukemia. American Journal of Medical Sciences. 1906;131:100–107.
- Yates JW, Wallace HJ, Ellison RR, Holland JF. Cytosine arabinoside (NSC-63878) and daunorubicin (NSC-83142) therapy in acute nonlymphocytic leukemia. Cancer Chemother Rep. 1973;57(4):485–488.
- Rowley JD. Identificaton of a translocation with quinacrine fluorescence in a patient with acute leukemia. Nature. 1973;243:290–293.
- Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the acute leukaemias. French–American–British (FAB) co-operative group. Br J Haematol. 1976;33(4):451–458.
- Estey E, Döhner H. Acute myeloid leukemia. Lancet. 2006;368(9550):1894–1907.
- Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood. 2008;111(5):2505–2515.
- Döhner H, Estey EH, Amadori S, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel. Blood. 2010;115(3):453–474.
- Perl AE, Martinelli G, Cortes JE, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N Engl J Med. 2019;381(18):1728–1740.
- Tallman MS, et al. Acute promyelocytic leukemia: evolving therapeutic paradigms. Blood. 2019;133(15):1630–1643.
- DiNardo CD, Pratz K, Pullarkat V, et al. Venetoclax combined with azacitidine, decitabine, or low-dose cytarabine in untreated patients with AML ineligible for intensive chemotherapy. N Engl J Med. 2020;383(7):617–629.
- Wei AH, Döhner H, Pocock C, et al. Oral azacitidine maintenance therapy for acute myeloid leukemia in first remission. N Engl J Med. 2020;383:2526–2537.
- DiNardo CD, Stein EM, Fathi AT, et al. Mutant IDH inhibitors in AML: clinical progress and future directions. Leukemia. 2021;35:681–692.
- Arber DA, Orazi A, Hasserjian RP, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemia: integrating morphologic, clinical, and genomic data. Leukemia. 2022;36:2413–2446.
- Döhner H, Wei AH, Appelbaum FR, et al. Diagnosis and management of AML in adults: 2022 ELN recommendations from an international expert panel. Blood. 2022;140(12):1345–1377.
- WHO Classification of Tumours Editorial Board. Haematolymphoid Tumours. WHO Classification of Tumours, 5th Edition, Volume 2. Lyon: IARC; 2022.
- Krivtsov AV, et al. Targeting menin in KMT2A-rearranged and NPM1-mutant acute myeloid leukemia: clinical development of menin inhibitors. Blood Cancer Discovery. 2023;4(2):95–107.