latincede; yaprak anlamına gelir.
fol asiti
(bkz: folik asit)
Folik asit
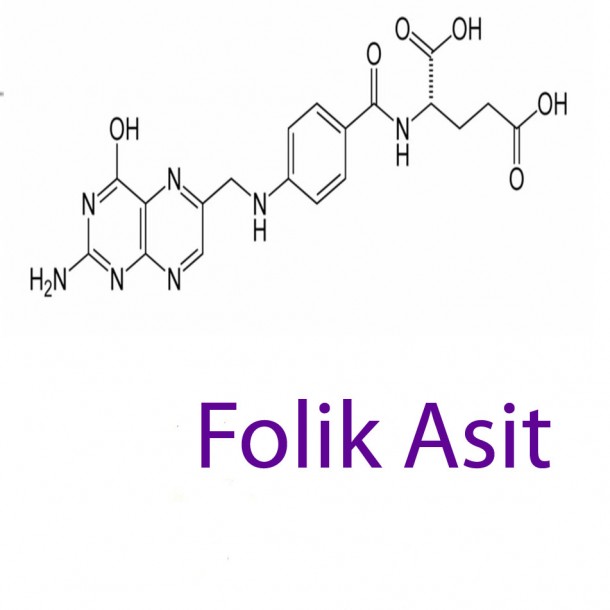
Folik asit ticari olarak tabletler biçiminde tek bir preparat olarak mevcuttur. Hem ilaç hem de besin takviyesi olarak satılmaktadır. Aynı zamanda kombine vitamin ve mineral takviyelerinde de mevcuttur. Folik asit adı yaprak olan Latince yapraktan türetilmiştir. Folik asit ilk olarak ıspanak yapraklarından izole edildi.
- latincede; folium (yaprak)’dan gelir. burdaki yaprak anlamı, yeşil bitkilerin yapraklarından gelmektedir.
- Mitchell ve arkadaşları bu vitamini, 1941 yılında ıspanak yapraklarında keşfettiler
Folik asit, DNA ve RNA sentezi gibi merkezi metabolik reaksiyonlarda yer alan B grubundan bir vitamindir (b9). Gebelik öncesi ve sırasında nöral tüp defektlerini önlemek, eksiklikleri önlemek ve megaloblastik anemiyi tedavi etmek için uygulanır. Endikasyona bağlı olarak, doz mikro- veya gram aralığındadır. Yan etkiler nadirdir ve genellikle sadece yüksek dozlarda ortaya çıkar. Bununla birlikte, çeşitli aktif bileşenlerle ilaç etkileşimleri mümkündür.
Kimya
Folik asit (C19H19N7O6, Mr = 441,4 g / mol), suda hemen hemen çözünmeyen, sarımsı ila turuncu renkli, kristal bir tozdur. Yapısal elementler olan pteridin, 4-aminobenzoik asit ve glutamik asitten oluşur. Folik asit, aktif tetrahidrofolatın (tetrahidrofolik asit, THF) bir ön ilacıdır.

Farmakoloji
Etkiler
Folik asit, merkezi metabolik reaksiyonlarda C1 moleküler yapı bloklarının transferinde bir koenzim olarak yer alır. Purinlerin, pirimidinlerin, nükleik asitlerin (DNA, RNA) sentezinde ve amino asitlerin metabolizmasında önemli rol oynar. Folik asit, DNA sentezi ve hücre yenilenmesi için gereklidir. Homosisteinin metiyonine parçalanmasında rol oynar. Yüksek homosistein seviyeleri çeşitli hastalıklarla ilişkilendirilmiştir.


Klinik
Endikasyon
- Hamilelik öncesinde ve sırasında nöral tüp defektlerinin birincil profilaksisi ve artan ihtiyaç nedeniyle, emzirme döneminde de takviye edilir.
- Folik asit eksikliğinden kaynaklanan megaloblastik anemiyi tedavi etmek için.
- Eksiklikleri önlemek için diyet takviyesi olarak.
- Düşük doz metotreksat tedavisi bağlamında, metotreksatla önceden doldurulmuş şırıngayla uygulama.

Uzman bilgilerine göre dozajlanır. Endikasyona bağlı olarak, doz mikrodan -miligrama kadar değişir.
DGE’ye göre, günlük ortalama folik asit gereksinimi:
| Yaş | Günlük ihtiyaç [µg] |
|---|---|
| < 4 ay | 60 |
| 4-12 ay | 85 |
| 1- 3 yıl | 120 |
| 4-6 yıl | 140 |
| 7-9 yıl | 180 |
| 10-12 yıl | 240 |
| 13-65 yıl | 300 |
Hamile ve emziren kadınların günlük ihtiyaçları artmaktadır ve bu ihtiyaç sırasıyla 550 µg ve 450 µg olarak verilmektedir.
Kontrendikasyon
Aşırı duyarlılık durumunda folik asit kontrendikedir. Zararlı anemi durumunda folik asit tek başına kullanılmamalıdır. B12 vitamini ile birlikte verilmelidir. İhtiyati tedbirlerin tamamı ürün bilgi sayfasında bulunabilir.
Etkileşimler
Diğerlerinin yanı sıra anti-epileptik ilaçlar, folik asit antagonistleri, florourasil, etanol ve kloramfenikol ile ilaç etkileşimleri mümkündür.
Yan etkileri
Folik asit genellikle iyi tolere edilir. Epilepside gastrointestinal bozukluklar, alerjik reaksiyonlar, psikiyatrik bozukluklar ve artmış nöbetler sadece miligram aralığında yüksek dozlarda ve daha uzun tedavi ile beklenebilir.
Diyet
- Gıdalarda, folik asit, birkaç glutamik asit kalıntısı ile mono- veya poliglutamat olarak çok sayıda kimyasal varyantta bulunur. Poliglutamatların biyoyararlanımı sentetik folik asitinkinden daha düşüktür.
- Gıdalardaki folik asitin % 25’i serbest haldedir ve ince bağırsakta kolaylıkla emilebilir. Folik asit içeren yiyecekler birkaç gün saklanır, yıkanır ve pişirilirse, folik asit içeriğinin üçte ikisine kadar kaybolur. Folik asit içeren yiyeceklere örnekler:
- ıspanak
- Kuşkonmaz
- Yaprak salataları
- Tahıl
- Ciğer

Tarih
- 1920’lerde bilim adamları folat eksikliği ve aneminin aynı durum olduğuna inanıyorlardı.
- 1931’de araştırmacı Lucy Wills, hamilelik sırasında anemiyi önlemek için gerekli besin maddesi olarak folatın tanımlanmasına yol açan önemli bir gözlem yaptı. Wills, aneminin bira mayası ile tersine çevrilebileceğini gösterdi.

- 1930’ların sonlarında, folat, bira mayasında düzeltici madde olarak tanımlandı.
- İlk olarak 1941’de Herschel K. Mitchell, Esmond E. Snell ve Roger J. Williams tarafından ıspanak yapraklarından ekstrakte edilerek izole edilmiştir.
- Tarihsel isimler arasında civcivlerde yapılan araştırmalardan sonra L. casei, faktör Bc vitamini ve maymunlarda yapılan araştırmalardan sonra M vitamini yer aldı.
- Bob Stokstad saf kristal formu 1943’te izole etti ve Amerikan Cyanamid Company’nin Lederle Laboratuvarlarında çalışırken kimyasal yapısını belirleyebildi.

- 1945’te saf kristal formda folik asit elde etmeye yönelik bu tarihsel araştırma projesi, Pearl, Lederle Lab’da Araştırma Direktörü Dr. Yellapragada Subbarow’un gözetimi ve rehberliği altında ‘folik asit çocukları’ adlı ekip tarafından yapıldı.

- Bu araştırma daha sonra 1948’de Sidney Farber tarafından çocukluk lösemisini tedavi etmek için kullanılan antifolat aminopterin sentezine yol açtı.
- 1950’lerde ve 1960’larda bilim adamları folat için biyokimyasal etki mekanizmalarını keşfetmeye başladılar. 1960 yılında, araştırmacılar folat eksikliğini nöral tüp defekti riskiyle ilişkilendirdiler.

- 1990’ların sonlarında, ABD ve Kanada hükümetleri, halk eğitim programlarına ve folik asit takviyelerinin mevcudiyetine rağmen, çocuk doğurma yaşındaki kadınların günlük folat tavsiyelerini karşılamada hâlâ bir zorluk olduğuna karar verdiler.
- Folat takviye programları. Aralık 2018 itibarıyla 62 ülke folik asit ile gıda zenginleştirmesini zorunlu kıldı.
poiesis
yunancada; yaradılış manasına gelir.
Hematopoez
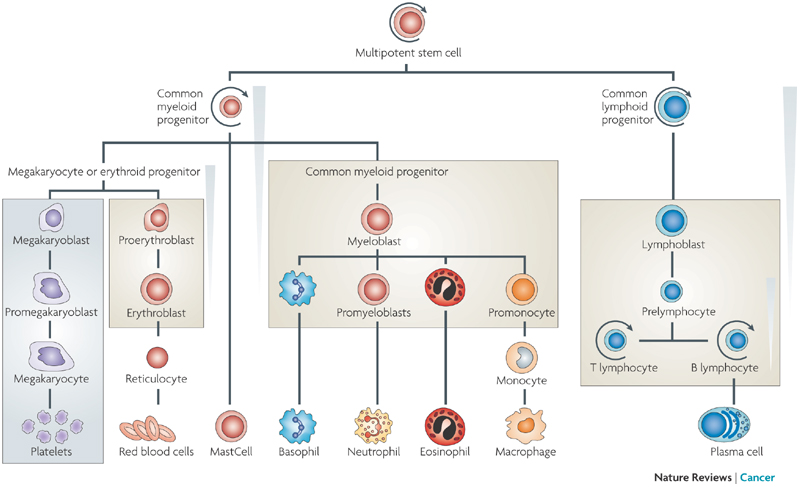
- Sinonim: Haematopoiesis, Hämatopoese, haematopoese, hematopoiesis, haemopoiesis veya hemopoiesis
- Yunancada; Kök hücrelerden kan hücrelerine dönüşümdür.(Bkz; Hemat-o-poez)
Eritrosit: yaklaşık 30–120 gün,
Trombosit: yaklaşık 3–10 gün
Kaynak: http://www.nature.com/nrc/journal/v8/n7/images/nrc2439-f3.jpg
Rett Sendromu

Rett sendromu, öncelikle kadınları etkileyen ve X kromozomundaki MECP2 genindeki mutasyonlarla bağlantılı olan nadir, ilerleyici bir nörogelişimsel bozukluktur. Yaygın gelişimsel bir bozukluk olarak ICD-10 kodu F84.2 altında sınıflandırılır. Aşağıda, Rett sendromunun mevcut anlayışının, epidemiyolojisinin, nedenlerinin, genetiğinin, klinik sunumunun, ilerlemesinin ve yönetiminin ayrıntılı bir dökümü, ek klinik bilgilere dayanarak çapraz kontrol edilmiş ve genişletilmiştir.

Epidemiyoloji:
- Rett sendromunun prevalansı yaklaşık olarak 10.000 ila 15.000 kadında 1’dir, ancak bazı kaynaklar 22.800’de 1 kadar düşük oranlar bildirmektedir. Bu tutarsızlık, tanı kriterlerindeki değişkenlik ve özellikle daha hafif veya atipik vakalarda eksik raporlama nedeniyle ortaya çıkmaktadır.
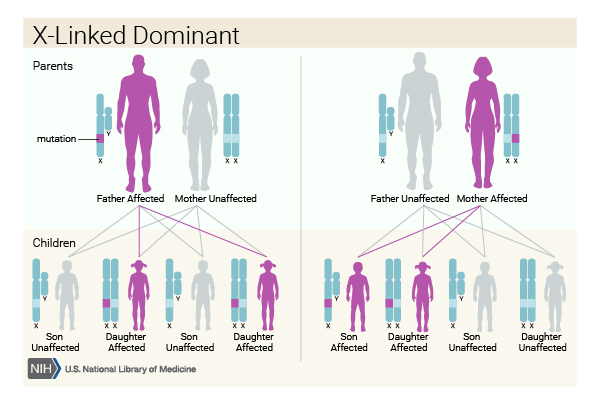
- Rett sendromu, X’e bağlı dominant kalıtım modeli nedeniyle ağırlıklı olarak kız çocuklarını etkiler. Bozukluk, tek bir X kromozomunda meydana geldiğinde mutasyonun etkilerinin ciddiyeti nedeniyle tipik olarak hayatta kalamayan erkek çocuklarda çok nadir görülür. Erkek vakaları son derece nadirdir ve genellikle somatik mozaiklik veya Klinefelter sendromu (47,XXY) içerir.
Tarihsel Arka Plan:
Andreas Rett tarafından ilk açıklama (1966):
- Avusturyalı çocuk doktoru Andreas Rett** sendromu ilk kez 1966 yılında tanımlamıştır. Bir grup kız çocuğunda gelişimsel gerileme, motor bozukluk ve karakteristik tekrarlayan el hareketleri gözlemlemiş ve bulgularını belgelemiştir. Bununla birlikte, çalışması nispeten belirsiz bir dergide Almanca olarak yayınlandı ve bu da daha geniş bilimsel topluluktaki ani etkisini sınırladı.
- Rett, el fonksiyon kaybı, “el yıkama” hareketleri, motor bozulma ve entelektüel gerileme gibi ayırt edici semptomları tanımlamıştır.

Bengt Hagberg (1983) tarafından **Uluslararası Tanınma:
- İsveçli çocuk doktoru Bengt Hagberg** 1983 yılında bu duruma uluslararası dikkat çekmiştir. Hagberg, meslektaşlarıyla birlikte, benzer semptomlar sergileyen 35 kızdan oluşan bir grubu analiz ederek Rett sendromunu ayrıntılı olarak tanımlayan ilk İngilizce makaleyi yayınladı.
- Hagberg’in yayını, Andreas Rett’in ardından bu durumu resmi olarak “Rett sendromu” olarak adlandıran ve tıp dünyasında geniş çapta tanınmasını sağlayan ilk yayın oldu. “Otizm, demans, ataksi ve kız çocuklarında amaca yönelik el kullanımının kaybından oluşan ilerleyici bir sendrom: Rett sendromu” başlıklı makale, sendromun anlaşılmasında bir dönüm noktası olmuştur.
Tanı Kriterlerinin Oluşturulması (1980’ler-1990’lar):
- 1980’ler ve 1990’lar** boyunca, araştırmacılar Rett sendromu için teşhis kriterlerini geliştirmeye başladılar. Bu zamana kadar, otizm spektrum bozuklukları kategorisi altında bir pervasif gelişimsel bozukluk olarak sınıflandırıldı (ICD-10: F84.2).
- Pediatristler ve nörologlar, Rett sendromunu diğer durumlardan ayıran karakteristik gelişimsel gerileme, motor disfonksiyon ve otistik benzeri davranışları tanımladılar.
MECP2 Gen Mutasyonunun Tanımlanması (1999):
- En önemli atılım 1999 yılında, Lübnan asıllı Amerikalı bir doktor ve genetikçi olan Huda Zoghbi, X kromozomu üzerindeki MECP2 genindeki mutasyonların Rett sendromu vakalarının çoğundan sorumlu olduğunu keşfettiğinde gerçekleşti. Bu keşif Nature dergisinde yayınlandı ve bozukluğun genetik temelinin anlaşılmasını değiştirdi.
- MECP2 (metil-CpG bağlayıcı protein 2)** beyin gelişiminde rol oynayan diğer genleri düzenleyen kritik bir gendir. MECP2’deki mutasyonlar normal nöronal işlevi bozarak Rett sendromunda görülen ilerleyici nörolojik gerilemeye yol açar.
Erkek Vakaları ve MECP2 Duplikasyonunu Anlamak (2000’lerin Başı):
- 2000’lerin başında**, daha fazla araştırma erkeklerde nadir görülen *Rett sendromu* vakalarını araştırdı ve MECP2 mutasyonları olan çoğu erkek hastanın bebeklik döneminde hayatta kalamadığını, bazılarının ise somatik mozaiklik veya Klinefelter sendromu gibi diğer kromozomal anormalliklerle hayatta kaldığını açıkladı.
- Ayrıca araştırmacılar, erkek çocuklarda Rett benzeri bir fenotiple sonuçlanan ve MECP2 geninin fazladan kopyalarını içeren bir durum olan MECP2 duplikasyon sendromunu keşfetmişlerdir.
Ek Genetik Nedenlerin Keşfi (CDKL5 ve FOXG1 Genleri, 2004-2010’lar):
- Çoğu Rett sendromu vakası MECP2’deki mutasyonlarla bağlantılı olsa da, daha sonraki çalışmalar CDKL5 (sikline bağımlı kinaz benzeri 5) ve FOXG1 (Forkhead box protein G1) gibi diğer genlerde atipik Rett sendromuna veya Rett benzeri semptomlara neden olabilen mutasyonları tanımlamıştır. Bu keşifler, bozukluğun genetik heterojenliğini vurgulamıştır.
- CDKL5 mutasyonları** daha erken nöbet başlangıcı ve daha ciddi gelişimsel gecikme ile ilişkiliyken, FOXG1 mutasyonları gelişimsel sorunların doğumdan itibaren belirgin olduğu konjenital Rett sendromu vakalarında yer almaktadır.
Hayvan Modellerinin Geliştirilmesi (2000’ler-2010’lar):
- MECP2 mutasyonunun keşfinin ardından araştırmacılar, bozukluğun patolojisini incelemek ve potansiyel tedavileri test etmek için Rett sendromunun fare modellerini geliştirdiler. Bu modeller, Rett sendromunun nörolojik semptomlarını taklit ederek hastalığın ilerleyişine dair içgörü sağlamıştır.
- Bu hayvan modelleri, klinik öncesi araştırmaların ilerletilmesinde etkili olmuş ve gen terapisi ve farmakolojik müdahalelerin test edilmesi için temel oluşturmuştur.
Gen Terapisi ve Farmakolojik Denemeler (2010’lar-Günümüz):
- Son yıllarda, gen terapisi Rett sendromunu tedavi etmek için umut verici bir yol olarak ortaya çıkmıştır. Bilim insanları, kusurlu MECP2 genini değiştirmenin veya onarmanın yollarını araştırmaktadır. Hayvan modellerinde gen terapisi kullanan erken preklinik çalışmalar, Rett benzeri semptomları tersine çevirme potansiyelini göstermiştir.
- gibi ilaçlar için klinik denemeler
Gen Tedavisi ve Farmakolojik Denemeler (2010’lar-Günümüz):
- Son yıllarda, gen terapisi Rett sendromunun tedavisi için umut verici bir yol olarak ortaya çıkmıştır. Bilim insanları, kusurlu MECP2 genini değiştirmenin veya onarmanın yollarını araştırmaktadır. Hayvan modellerinde gen terapisi kullanan erken klinik öncesi çalışmalar, Rett benzeri semptomları tersine çevirme potansiyelini göstermiştir.
- Rett sendromu hastalarında apne gibi solunum sorunlarını tedavi etmeyi amaçlayan sarizotan gibi ilaçlar ve MECP2 eksikliğinin aşağı akış etkilerini hedefleyen diğer bileşikler için klinik denemeler halen devam etmektedir.
Uluslararası Rett Sendromu Vakfı ve Savunuculuk (2000’ler-Günümüz):
- Uluslararası Rett Sendromu Vakfı (IRSF)** gibi kuruluşların kurulması, farkındalığın artırılmasında, araştırmaların finanse edilmesinde ve Rett sendromundan etkilenen ailelerin desteklenmesinde çok önemli bir rol oynamıştır.
- Küresel farkındalık kampanyaları ve hasta savunuculuğu, bilim insanları, bakıcılar ve klinisyenler arasında araştırma finansmanının ve işbirliğinin artmasına yol açarak Rett sendromu tedavilerine yönelik araştırmaları ilerletmiştir.
Nedenleri:
- Vakaların % 90’ından fazlasında Rett sendromuna MECP2 (metil-CpG bağlayıcı protein 2) genindeki mutasyonlar neden olur. MECP2, metillenmiş DNA’ya bağlanarak ve gen ekspresyonunu kontrol ederek diğer genlerin düzenlenmesinde rol oynadığı için nöronal fonksiyon için çok önemlidir.
- MECP2 duplikasyonları** Rett sendromu vakalarının küçük bir alt kümesini oluşturur. Nadir durumlarda, CDKL5 (siklin bağımlı kinaz benzeri 5) veya FOXG1 (Forkhead box protein G1) gibi diğer genlerdeki mutasyonlar Rett benzeri semptomlarla ilişkilidir.
- Bu mutasyonlar tipik olarak de novo yani sperm veya yumurta oluşumu sırasında kendiliğinden meydana gelir ve ebeveynlerden miras alınmaz.
Genetik Kalıtım:
- Rett sendromu X’e bağlı baskın kalıtım modelini takip eder. Çoğu durumda, etkilenen bireyler mutasyonu ebeveynlerinden ziyade de novo olarak miras alırlar.
- Kadınlar** tipik olarak, normal X kromozomu tarafından bir miktar telafiye izin veren iki X kromozomunun varlığı nedeniyle etkilenir. Erkeklerde** sadece bir X kromozomuna sahip olmak, MECP2 mutasyonunun ciddi gelişimsel sorunlara neden olabileceği ve genellikle doğum öncesi veya doğum sonrası erken ölüme yol açabileceği anlamına gelir. Bununla birlikte, tipik olarak somatik mozaizm (sadece bazı hücrelerin mutasyonu taşıdığı) veya Klinefelter sendromunda (47,XXY) görüldüğü gibi ekstra bir X kromozomu içeren nadir Rett sendromlu erkek vakaları mevcuttur.
Semptomlar:
Rett sendromu, tipik olarak gelişimin çeşitli aşamalarında ilerleyen çok çeşitli nörolojik ve motor semptomlara sahiptir. Temel özellikler şunları içerir:
- El stereotipleri, özellikle karakteristik ve tanısal olan tekrarlayan el sıkma veya “yıkama” hareketleri.
- Amaca yönelik el kullanımı, konuşma ve motor beceriler dahil olmak üzere edinilmiş becerilerin** kaybı.
- Sosyal etkileşime olan ilgi genellikle devam etse de, sosyal geri çekilme ve göz temasının azalması gibi Otistik benzeri davranışlar.
- Ciddi bilişsel bozukluk**: Entelektüel gelişim önemli ölçüde bozulur ve bunamaya benzer bir duruma yol açar.
- Mikrosefali (baş büyümesinin azalması), görünüşte normal bir gelişim döneminden sonra fark edilir hale gelir.
- Nöbetler**: Rett sendromlu birçok çocukta *epilepsi* gelişir.
- Spastisite ve diğer hareket bozukluklarının yanı sıra Apraksi (motor fonksiyonları yerine getirememe) ve ataksi (koordine olmayan hareketler) yaygındır.
- Hiperventilasyon, aerofaji** (hava yutma) ve apne (solunumun durduğu dönemler) dahil olmak üzere nefes alma düzensizlikleri.
- Genellikle PEG (perkütan endoskopik gastrostomi) tüpüyle besleme gibi tıbbi müdahaleler gerektiren kabızlık ve yutma güçlükleri gibi gastrointestinal sorunlar.
Hastalığın Seyri ve Aşamaları:
Rett sendromunun ilerlemesi, Hagberg ve Witt-Engerström’ün çalışmalarına dayanarak tipik olarak dört aşamaya ayrılır:
Aşama 1: Erken Başlangıç (Yavaşlama Aşaması, 6-18 ay):
- İlk gelişim normal görünür, ancak ince işaretler ortaya çıkmaya başlar. Bunlar arasında daha yavaş motor gelişim, çevreye ilgide azalma ve sosyal katılımda azalma yer alır. Çocuğun baş çevresi tipik büyüme modellerinin gerisinde kalmaya başlar (mikrosefaliye yol açar).
2. Evre: Hızlı Yıkıcı Evre (1-4 yaş):
- Bu, el kullanımı ve dil başta olmak üzere edinilmiş becerilerin kaybı ile belirgindir. El yıkama stereotipleri ve tekrarlayan hareketler, artan sosyal geri çekilme ve otistik benzeri davranışlar ile birlikte belirgin hale gelir. Çığlık atma** ve sinirlilik dönemleri ortaya çıkabilir.
3. Aşama: Plato (Kararlı Aşama, 2-10 yıl):
- Bu evrede gerileme yavaşlar ve bazı sosyal etkileşim ve iletişim becerileri hafifçe iyileşebilir. Bununla birlikte, epileptik nöbetler, apraksi ve motor defisitler (örneğin, dengesiz yürüyüş) genellikle kötüleşir veya daha belirgin hale gelir.
Evre 4: Geç Motor Bozulma (10 yıl sonra):
- Kaba motor beceriler bozulmaya devam ederek ciddi motor bozukluklara yol açar. Hastaların çoğu tekerlekli sandalyeye bağlı hale gelir. Hareket kabiliyetindeki azalmaya rağmen nöbetler azalabilir ve sosyal ve bilişsel beceriler stabilize olabilir, hatta iyileşebilir. Bununla birlikte, hareket daha katı hale gelir ve kas erimesi yaygındır.
Tedavi ve Yönetim:
Şu anda Rett sendromu için tedavi yok ve tedavi öncelikle semptomları yönetmeye ve yaşam kalitesini iyileştirmeye odaklanıyor. Temel müdahaleler şunları içerir:
Terapiler:
- Hareketliliği sürdürmek, kas gücünü artırmak ve amaca yönelik el kullanımını teşvik etmek için fizik tedavi ve meslek terapisi.
- İletişim becerilerini geliştirmek için konuşma terapisi, ancak birçok birey sözel bozukluklar nedeniyle alternatif iletişim yöntemlerine güvenecektir.
- Müzik terapisi** ve hipoterapi (atlarla terapi) ruh halini, motor koordinasyonu ve sosyal katılımı iyileştirmede faydalar göstermiştir.
Farmakolojik Müdahaleler:
- Antiepileptik ilaçlar (AED’ler)** genellikle nöbetleri yönetmek için kullanılır.
- apne** gibi solunum sorunları için sarizotan gibi ilaçlar klinik çalışmalarda araştırılmaktadır. Sarizotan, solunum anormalliklerini hafifletmek için tasarlanmıştır ve erken çalışmalarda umut vaat ettiğini göstermiştir.
Beslenme Desteği:
- Ciddi yutma güçlüklerinde, beslenme sağlamak ve aspirasyon pnömonisi riskini azaltmak için bir PEG tüpü kullanılabilir.
Spastisite ve Hareket Bozukluklarının Yönetimi:
- Fizyoterapi**, *plintleme* ve baklofen veya botulinum toksin enjeksiyonları gibi ilaçlar spastisite ve motor disfonksiyonun yönetilmesine yardımcı olabilir.
Gastrointestinal ve Solunum Sorunlarının İzlenmesi ve Tedavisi:
- Yutma bozuklukları**, *kabızlık* ve solunum anormallikleri gastrostomi ile beslenme ve solunum düzeninin izlenmesi gibi müdahalelerle sürekli yönetim gerektirir.
Gelecekteki Yönelimler:
Altta yatan MECP2 işlev bozukluğunu hedef alan gen tedavisi ve farmakolojik müdahaleler üzerine araştırmalar devam etmektedir. Deneysel tedaviler MECP2 işlevini geri kazandırmayı veya eksikliğinin aşağı yönlü etkilerini hafifletmeyi amaçlamakta ve gelecekte daha etkili tedaviler için umut vermektedir.
İleri Okuma
- Rett, A. (1966). “On a remarkable syndrome of cerebral atrophy associated with hyperammonemia in childhood.” Wiener Medizinische Wochenschrift, 116(37), 723-726.
- Hagberg, B., et al. (1983). “A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: report of 35 cases.” Annals of Neurology, 14(4), 471-479.
- Amir, R. E., et al. (1999). “Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2.” Nature Genetics, 23(2), 185-188.
- Chahrour, M., et al. (2008). “MeCP2, a key contributor to neurological disease, activates and represses transcription.” Science, 320(5880), 1224-1229.
- Neul, J. L., et al. (2010). “Rett syndrome: Revised diagnostic criteria and nomenclature.” Annals of Neurology, 68(6), 944-950.
statik
yunancada; duraklamak, tutmak anlamına gelir.
Bkz; stasis
Fonksiyonel manyetik rezonans görüntüleme
Fonksiyonel manyetik rezonans tomografisi, fizikçi Kenneth Kwong‘a kadar uzanan manyetik rezonans tomografisinin bir çeşididir. Yöntem, aktif sinir hücrelerinin enerji gereksinimlerinin neden olduğu çeşitli beyin bölgelerindeki doku kan akışındaki (rCBF) değişiklikleri ölçer. Böylelikle beyin dokusundaki fonksiyonel süreçleri kesitsel görüntüler dizisi şeklinde temsil edebilir.
Kinematik manyetik rezonans tomografi ayrıca ‘fonksiyonel manyetik rezonans tomografi’ terimi altında sınıflandırılır.

Temeli
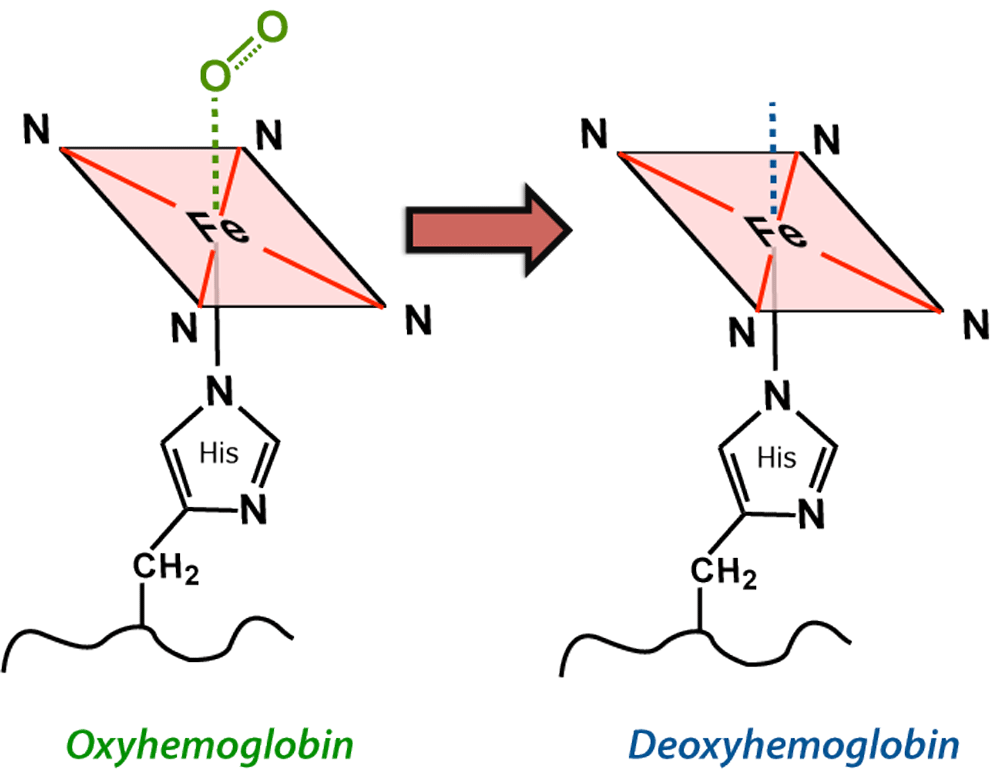
- FMRI temsilinin temeli, sinyal tespiti için oksijenden zengin ve oksijenden fakir kanın farklı manyetik özelliklerini kullanan, daha kesin olarak oksihemoglobin ve deoksihemoglobin arasındaki farkı kullanan sözde BOLD etkisidir.
- Oksihemoglobin diyamanyetiktir ve çevresindeki dokunun manyetik özellikleri üzerinde hiçbir etkisi yoktur. Öte yandan deoksihemoglobin paramanyetiktir. Bu özellik, ayrı fakat gösterilebilir manyetik alan değişikliklerine yol açar.
- Beyin bölgeleri uyarıldığında, metabolizmada sınırlı bir artış olur ve bu, artan beyin kan akışıyla bölgesel olarak kendini gösterir. Bu, oksijenli ile oksijeni giderilmiş hemoglobin oranını değiştirir ve bu da bir sinyal değişikliğine neden olur. Kayıtlar zaman içinde iki farklı noktada yapılırsa (dinlenme durumuna karşı uyarılmış durum), istatistiksel test yöntemleri kullanılarak birbirleriyle karşılaştırılabilir. Uyarılmış alanlar daha sonra bilgisayar tarafından uzaysal olarak atanır ve görsel olarak görüntülenir.
Çıkarım
FMRI, sinir hücresi aktivitesini birkaç saniyelik bir zaman penceresinde milimetre hassasiyetinde lokalize edebilir. Beynin işlevsel süreçlerine, özellikle bilişsel ve duygusal süreçlerin topografyasına yeni bakış açıları sağlar. Verileri değerlendirirken, beyin dokusunda hemodinamikteki değişimin doğrudan sinirsel aktivite ile değil, sadece birkaç saniyelik bir gecikme süresinden sonra meydana geldiğine dikkat edilmelidir.
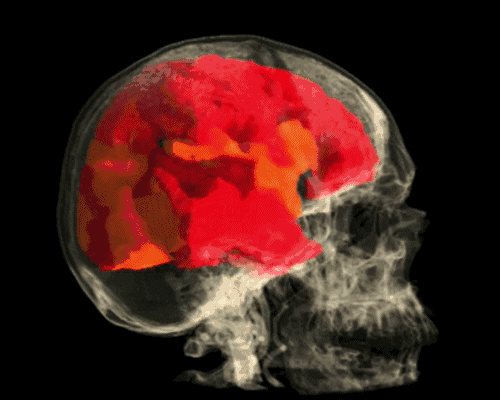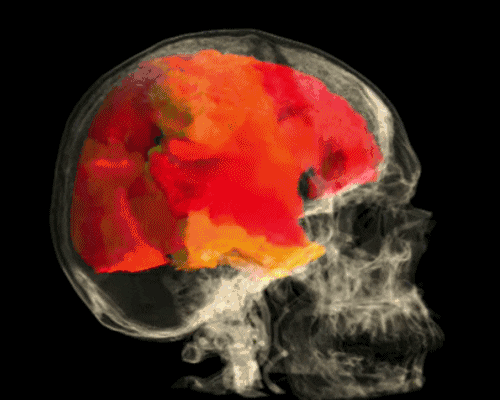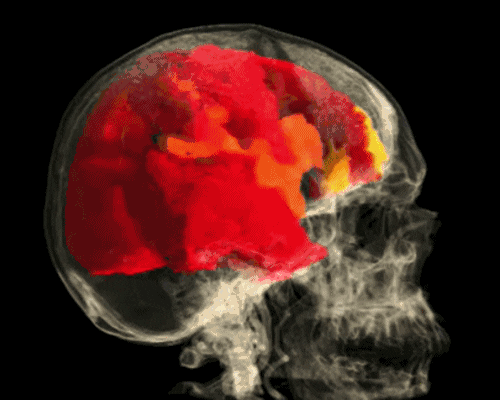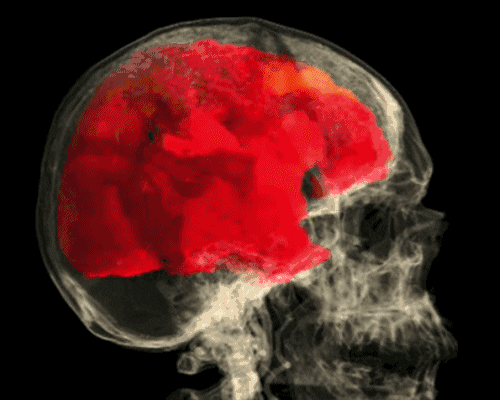
Uygulama alanları
- FMRI esas olarak nöroradyolojide preoperatif açıklama için kullanılır. Örneğin, onların yardımı ile dil lateralizasyonu belirlenebilir. Bu, Wernicke bölgesine veya Broca bölgesine yakın planlı bir beyin tümörü rezeksiyonu için önemlidir, böylece önemli beyin bölgeleri korunabilir. Terapiye dirençli temporal lob epilepsileri ve nöroşirürji tedavisi gerektiren paryetal lob epilepsilerinde de büyük önem taşımaktadır. Bu amaçla, hastanın aynadan bakabileceği bir monitör aracılığıyla hastaya çeşitli görevler veya parametreler sunulur:
- Fiil üretme parametreleri: İsimler hastaya 30 saniyelik aralıklarla her biri 5 dakikalık bir süre boyunca 30 saniye süreyle sunulur. Şimdi her isim için uygun fiiller düşünmelidir. Örnek: ‘top’ kelimesi sunulur. Örneğin, hasta ‘oynamayı’, ‘yakalamayı’ veya ‘fırlatmayı’ düşünebilir. Bu parametre öncelikle Broca’nın konuşma-motor alanını uyarır.
- Cümleler Parametresi: Cümleler hastaya yukarıdaki ile aynı şekilde sunulur. Cümlenin içerik olarak anlamlı olup olmadığını düşünmelidir. Örnek: ‘Futbolcu bir topa vuruyor.’ İçerik doğru. Fırıncı araba pişiriyor. İçerik doğru değil. Bu parametre esas olarak dil-duyusal Wernicke bölgesini uyarır. Bu parametre ayrıca görsel kortekste daha güçlü aktivasyonlara yol açar.
- Nesne adlandırma parametreleri: Bu parametre ile hastaya, ilk iki parametreyle aynı şekilde adlandırması gereken nesnelerin resimleri ve çizimleri gösterilir. Bu parametre aynı zamanda özellikle Broca bölgesini uyarır. Görsel korteks ayrıca burada fiil üretme parametresindekinden daha güçlü bir şekilde etkinleştirilir.
- Daha önce bahsedilen endikasyonlara ek olarak, fMRI aynı zamanda beyin araştırmaları, psikiyatri ve nöropazarlamada da artan bir ilgi görüyor.

MCHC
- ingilizcede; mean corpuscular haemoglobin concentration demektir.
- bir erythrocyt deki hemoglobin derişimini ifade eder.
- mchc = mch / mcv
gone
yunancada(f);cinsiyet.
-üreme hücreleri.