Farmakokinetikte permeasyon, bir ilacın hücre zarından geçişini ifade eder. Bu terim genellikle sindirim sistemindeki bağırsak emilimi ile bağlantılı olarak kullanılır. Bu süreçte, aktif bileşen molekülleri dozaj formundan salındıktan sonra bağırsak lümeninden enterositlerin (bağırsak hücreleri) hücre zarlarından kan dolaşımına geçer. Bununla birlikte, permeasyon tüm farmakokinetik süreçlerde merkezi bir rol oynamaktadır.
Aktif maddeler öncelikle pasif difüzyonla, taşıyıcılar, kanallar ve endositoz yoluyla ve membranlar boyunca veziküllerle ekzositoz yoluyla kan dolaşımına ulaşır. Paraselüler taşıma, hücreleri geçen yolu ifade eder.
Geçirgenliği etkileyen faktörler şunlardır:
İlacın fizikokimyasal özellikleri, örneğin lipofiliklik, polarite, yük.
Pitavastatin, tamamen sentetik, güçlü bir 3-hidroksi-3-metilglutaril-koenzim A (HMG-CoA) redüktaz inhibitörü olup, statinler sınıfına ait lipid düşürücü bir ajandır. Klinik kullanımı, primer hiperkolesterolemi ve kombine (mixed) dislipidemi tedavisinde, LDL-kolesterol (LDL-K) ve total kolesterolü belirgin biçimde düşürmesi ve HDL-kolesterolü (HDL-K) görece güçlü yükseltme kapasitesi etrafında şekillenmiştir.
Farmakodinamik ve farmakokinetik özellikleri dikkate alındığında pitavastatin, “üçüncü kuşak” veya “yüksek potent” statinler arasında konumlandırılır: lipofilik bir molekül olup karaciğer hedeflenmesi OATP aracılı alım üzerinden olur, CYP3A4 bağımlılığı minimaldir ve eliminasyon yarı ömrü yaklaşık 11–12 saattir.
2. Etimoloji ve Adlandırma
“Pitavastatin” adı, statin sınıfı ilaçların ortak adlandırma şemasını takip eder:
Son ek “-vastatin”, HMG-CoA redüktaz inhibitörlerini işaret eden, sınıf içinde yaygın kullanılan bir unsurdur (lovastatin, simvastatin, atorvastatin, rosuvastatin vb.).
Baş kısım olan “pita-” için literatürde resmi bir açıklama bulunmamakla birlikte, molekülün özgün kimyasal mimarisine (kinolin çekirdeği, siklopropil yan zincir ve lipofilik aromatik yapı) ve firma içi kod adlandırmalarına (NK-104 gibi) dayanan sentetik bir kök olduğu kabul edilir.
Bu çerçevede pitavastatin, kimyasal yapı temelli rasyonel ilaç tasarımının ürünü olan ve adı da bu yapı ve sınıf aidiyetini yansıtan bir molekül olarak görülebilir.
3. Tarihçe ve Geliştirme Süreci
Pitavastatin ilk olarak Japonya’da Nissan Chemical Industries tarafından keşfedilmiş, daha sonra Kowa Pharmaceuticals tarafından geliştirilmiştir.
Zaman çizelgesi kabaca şu şekildedir:
1987 – Pitavastatin için ilk patent başvuruları.
2003 – Japonya’da tıbbi kullanım için onay ve lansman.
2009 – ABD Gıda ve İlaç Dairesi (FDA) tarafından Livalo® (pitavastatin kalsiyum) için ilk onay (3 Ağustos 2009).
2010 – Birleşik Krallık İlaç ve Sağlık Ürünleri Düzenleme Kurumu (MHRA) referans üye ülke olarak Avrupa Birliği’nde Livazo®/Alipza® için olumlu değerlendirme, ardından çok sayıda AB ülkesinde ulusal onaylar.
2017 – ABD’de pitavastatin magnezyum tuzu (Zypitamag®) için ek onay, farmasötik eşdeğer olarak kabul edilir.
Bugün pitavastatin Asya, Avrupa, Latin Amerika ve Kuzey Amerika’da, başlıca Livalo®, Livazo®, Zypitamag® gibi ticari isimler altında, ayrıca çok sayıda jenerik form ile pazarlanmaktadır.
4. Kimyasal Yapı, Fizikokimyasal Özellikler ve Yapı-Aktivite İlişkisi
4.1. Kimyasal formül ve temel özellikler
Pitavastatinin serbest asit formunun molekül formülü C₂₅H₂₄FNO₄, ortalama molekül ağırlığı yaklaşık 421,5 g/mol’dür.
Klinik kullanıma giren form pitavastatin kalsiyum (hemikalsiyum tuzu) olup bunun molekül formülü C₅₀H₄₆CaF₂N₂O₈’dir. Pitavastatin kalsiyum:
Beyaz–açık sarı, kokusuz, ışığa duyarlı ve hafif higroskopik bir tozdur.
Suda çok az çözünür; lipofilik çözücülerde ve bazı organik araçlarda daha iyi çözünür.
Bu düşük suda çözünürlüğe rağmen, uygun tuz formu ve formulasyon sayesinde oral biyoyararlanımı klinik açıdan anlamlı düzeyde kalır.
4.2. Yapısal özellikler
Pitavastatinin IUPAC adı (serbest asit için) kabaca şu şekildedir:
Dihidroksi heptanoik asit yan zinciri: Tüm statinlerde olduğu gibi, HMG-CoA redüktazın doğal substratı mevalonata yapısal olarak benzeyen bu yan zincir, enzimin aktif bölgesine yüksek afiniteyle bağlanmayı sağlar.
Kinolin çekirdeği: Aromatik, planarımsı bu halka sistemi, lipofilikliği ve hedeflenmiş karaciğer alımını destekler; farmakoforun şekillenmesinde kritik rol oynar.
Siklopropil grubu ve 4-fluorofenil substitüenti: HMG-CoA redüktaz ile hidrofobik etkileşimleri güçlendirirken, molekülün metabolik stabilitesini ve reseptör bağlanma özelliklerini modüle eder.
Pitavastatin, birçok başka statinin aksine bir ön ilaç (prodrug) değildir; uygulanan formu farmakolojik olarak aktiftir ve metabolik aktivasyon gerektirmez.
5. Farmakodinamik Özellikler
5.1. HMG-CoA redüktaz inhibisyonu
Pitavastatin, hepatik HMG-CoA redüktazı kompetitif ve geri dönüşümlü olarak inhibe eder. Enzimin katalize ettiği reaksiyon, HMG-CoA’dan mevalonata dönüşümü olup, kolesterol biyosentezinin hız sınırlayıcı basamağıdır.
In vitro veriler, pitavastatinin HMG-CoA redüktaz için nanomolar düzeyde (IC₅₀ ≈ 6–7 nmol/L) potent bir inhibitör olduğunu göstermiştir.
5.2. LDL reseptör regülasyonu ve lipid profili
HMG-CoA redüktaz inhibisyonu hepatik kolesterol havuzunu azaltır. Bunun sonucunda:
Dolaşımdaki LDL partiküllerinin karaciğer tarafından temizlenmesi hızlanır,
Plazma LDL-K konsantrasyonu belirgin şekilde azalır.
Klinik çalışmalarda:
2 mg/gün pitavastatin ile ortalama LDL-K düşüşü yaklaşık %39,
4 mg/gün ile yaklaşık %45 düzeyindedir.
Pitavastatin ayrıca:
Total kolesterol ve trigliseritleri düşürür,
HDL-K üzerinde belirgin ve zamanla artan bir yükseltici etki gösterebilir; uzun süreli gözlemsel çalışmalarda özellikle düşük başlangıç HDL-K düzeyine sahip hastalarda %20–25’e varan artışlar bildirilmiştir.
5.3. Pleiotropik etkiler
Statin sınıfının tipik pleiotropik etkileri pitavastatin için de gösterilmiştir:
Endotelyal nitrik oksit sentezinin artışı ve endotelyal fonksiyonun iyileşmesi,
İnflamatuvar sitokinlerin ve C-reaktif proteinin azalması,
Oksidatif stres belirteçlerinde azalma ve aterosklerotik plak stabilitesinin artışı,
Vasküler yeniden şekillenme süreçlerinin baskılanması.
Bu etkiler, LDL-K düşüşünden bağımsız kardiyovasküler yararlara katkıda bulunabilecek ek mekanizmalar olarak değerlendirilir.
6. Farmakokinetik Özellikler
6.1. Emilim ve biyoyararlanım
Pitavastatin oral alımdan sonra gastrointestinal sistemden hızla emilir:
Maksimum plazma konsantrasyonu (Cmax) genellikle ≈ 1 saat içinde ulaşılır.
Mutlak biyoyararlanım yaklaşık %51–60 düzeyindedir.
Yağdan zengin bir öğün Cmax’i anlamlı ölçüde azaltabilir, ancak AUC (toplam maruziyet) belirgin olarak değişmez; klinik önem sınırlı kabul edilir.
Bu özellikler nedeniyle pitavastatin öğünlerden bağımsız kullanılabilir.
6.2. Dağılım
Pitavastatin:
Plazma proteinlerine >%99 oranında bağlanır (özellikle albumin ve α₁-asit glikoprotein).
Ortalama dağılım hacmi yaklaşık 130–150 L düzeyindedir; bu da ekstravasküler alana anlamlı dağılımı ve karaciğer hedeflenmesini destekler.
6.3. Metabolizma
Pitavastatinin temel özelliklerinden biri, CYP450 sistemine bağımlılığının düşük olmasıdır:
Primer metabolizma yolu hepatik glukuronidasyondur; başlıca UGT1A3 ve UGT2B7 (ayrıca UGT1A1) enzimleriyle pitavastatin-O-glukuronid oluşur ve bu metabolit hızla lakton formuna siklize olur.
CYP450 metabolizması minimaldir; daha çok CYP2C9 ve CYP2C8 küçük katkılar sağlar, CYP3A4 neredeyse hiç rol oynamaz.
Bu metabolik profil, CYP3A4 üzerinden yoğun ilaç–ilaç etkileşimi potansiyeline sahip diğer statinlere (ör. simvastatin, atorvastatin) göre pitavastatini avantajlı kılar.
6.4. Eliminasyon
Ortalama plazma eliminasyon yarı ömrü ≈ 11–12 saattir.
Radyoaktif işaretli çalışmalar, verilen dozun yaklaşık %79’unun feçesle, ≈%15’inin idrarla atıldığını göstermiştir; safra yoluyla hepatobiliyer atılım baskındır.
6.5. Özel popülasyonlar
Yaşlı hastalar (>65 yaş): Farmakokinetik profil genç erişkinlerle benzerdir; özel doz ayarlaması tipik olarak gerekli değildir.
Irksal farklılıklar: Bazı verilerde siyah/Afro-Amerikan gönüllülerde Cmax ve AUC’nin beyaz gönüllülere göre hafifçe daha düşük olduğu bildirilmiştir; klinik olarak anlamlı bir doz ayarlama gerekliliği ortaya konmamıştır.
Böbrek yetmezliği: Orta–ağır böbrek yetmezliğinde ve hemodiyaliz hastalarında maruziyet artar; bu nedenle tavsiye edilen maksimum doz daha düşüktür (aşağıda doz bölümünde ayrıntılandırılmıştır).
7. Terapötik Endikasyonlar ve Klinik Etkililik
Pitavastatin, kılavuz ve ruhsat bilgilerine göre aşağıdaki durumlarda diyete ek tedavi olarak endikedir:
Erişkinlerde primer hiperkolesterolemi (heterozigot ailesel hiperkolesterolemi dahil),
Kombine (mixed) dislipidemi,
ABD etiketinde ayrıca, 8 yaş ve üzeri heterozigot ailesel hiperkolesterolemili çocuklarda LDL-K ve ApoB düşürülmesi.
Randomize kontrollü ve uzun dönem gözlemsel çalışmalarda pitavastatin:
Non-HDL-K, ApoB gibi aterojenik lipoprotein belirteçlerini azaltmaktadır.
Glikoz metabolizması üzerine nötr veya nispeten daha elverişli etkileri olduğuna dair veriler, diyabet veya metabolik sendromu olan ve yüksek kardiyometabolik risk taşıyan hasta gruplarında pitavastatine özel ilgi doğurmuştur.
8. Dozaj ve Uygulama
Pitavastatin, film kaplı tabletler halinde 1 mg, 2 mg ve 4 mg güçlerinde kullanıma sunulur.
8.1. Erişkin dozajı
Standart başlangıç dozu: 2 mg günde bir kez, oral.
Doz aralığı: 1–4 mg günde bir kez.
Maksimum doz: 4 mg/gün.
Tabletler:
Günde bir kez,
Öğünlerden bağımsız olarak,
Her gün aynı saatte alınmalıdır.
Lipidler genellikle tedavi başlangıcından veya doz değişikliğinden en erken 4 hafta sonra yeniden değerlendirilir; hedef LDL-K düzeyine göre doz titrasyonu yapılır.
8.2. Pediatrik kullanım
8 yaş ve üzerindeki heterozigot ailesel hiperkolesterolemili çocuklarda önerilen başlangıç dozu genellikle 2 mg/gün, maksimum 4 mg/gün’dür.
Daha küçük yaş gruplarında güvenlilik ve etkinlik verileri sınırlıdır; kullanım ruhsata ve kılavuzlara göre değerlendirilmelidir.
8.3. Böbrek yetmezliğinde doz ayarlaması
Orta–ağır böbrek yetmezliği (eGFR 15–59 mL/dk/1,73 m²) veya son dönem böbrek yetmezliği ve hemodiyaliz:
Başlangıç: 1 mg/gün,
Maksimum: 2 mg/gün.
8.4. Karaciğer yetmezliği
Aktif karaciğer hastalığı veya açıklanamayan kalıcı transaminaz yüksekliği olan hastalarda kontrendikedir.
Hafif–orta derecede karaciğer bozukluğunda, bazı AB ürün bilgilerinde 4 mg doz önerilmez, 2 mg/gün’ün üzerinde çıkılmaması ve sık izlem vurgulanır.
9. Kontrendikasyonlar
Pitavastatin şu durumlarda kullanılmamalıdır:
Pitavastatin veya diğer statinlere ya da preparattaki yardımcı maddelere karşı aşırı duyarlılık,
Ciddi karaciğer yetmezliği veya aktif karaciğer hastalığı,
Serum transaminazlarında üst normal sınırın 3 katından fazla, açıklanamayan kalıcı artış,
Myopati öyküsü veya mevcut kas hastalığı,
Hamilelik ve emzirme,
Çocuk doğurma potansiyeli olan, yeterli kontrasepsiyon kullanmayan kadınlar,
Siklosporin ile birlikte kullanım (AB SmPC’de açık kontrendikasyon).
10. İlaç-İlaç Etkileşimleri
Pitavastatin, başlıca hepatik organik anyon taşıyıcı polipeptitlerin (OATP1B1, OATP1B3) substratıdır; bu taşıyıcıları inhibe eden veya onlar üzerinden taşınan ilaçlarla anlamlı farmakokinetik etkileşimlere girebilir.
Önemli etkileşimler:
Siklosporin: OATP inhibisyonu ile pitavastatin AUC’sinde yaklaşık 4–5 kat artış; kombinasyon kontrendikedir.
Eritromisin ve diğer makrolidler: Pitavastatin maruziyetini 2–3 kat arttırabilir; bazı kılavuzlarda tedavi süresince pitavastatin ara verilmesi, ABD etiketinde ise en fazla 1 mg/gün ile sınırlandırılması önerilir.
Gemfibrozil ve diğer fibratlar: AUC’de hafif–orta artış; sınıf etkisi olarak myopati/rabdomiyoliz riskini yükseltir; dikkatli kullanılmalı, mümkünse alternatif lipid düşürücü kombinasyonlar tercih edilmelidir.
Niasin (yüksek doz) ve fusidik asit: Statin-ilişkili kas toksisitesini artırabilecek kombinasyonlar; fusidik asit ile kombinasyonda ciddi rabdomiyoliz olguları bildirildiği için pitavastatine geçici ara verilmesi önerilir.
HIV proteaz inhibitörleri (atazanavir, darunavir/ritonavir, lopinavir/ritonavir): Farmakokinetik düzeyde değişiklikler olsa da çoğu durumda klinik olarak yönetilebilir; doz ayarlama gerekliliği molekül bazında değerlendirilmelidir.
Varfarin: Pitavastatin 4 mg ile birlikte varfarinin farmakokinetiği genellikle anlamlı değişmez; yine de başlangıçta ve doz değişikliklerinde INR izlemi önerilir.
CYP450 sistemi üzerinden etkileşim potansiyeli, özellikle CYP3A4 düzeyinde, diğer lipofilik statinlere kıyasla düşüktür; bu durum çoklu ilaç kullanan, polifarmasili yaşlı hastalarda pitavastatini farmakolojik olarak cazip kılar.
11. Advers Etkiler ve Güvenlilik Profili
11.1. Sık advers etkiler
Klinik çalışmalarda ve post-pazarlama gözlemlerinde en sık bildirilen yan etkiler çoğu statinle örtüşür:
Hafif ve genellikle geçici serum transaminaz yükselmeleri.
Bu yan etkiler çoğu zaman hafif–orta şiddettedir ve tedavinin sürdürülmesine engel olmaz.
11.2. Kas toksisitesi ve rabdomiyoliz
Statin sınıfına özgü kas toksisitesi riski pitavastatin için de geçerlidir:
Hafif miyaljiden laboratuvar olarak doğrulanmış miyopatiye (CK > 10× üst sınır) ve nadiren rabdomiyoliz ile seyreden, böbrek yetmezliğine ilerleyebilen ağır tablolara kadar spektrum söz konusudur.
Yüksek doz, ileri yaş, eşlik eden böbrek yetmezliği, hipotiroidi, kas hastalığı öyküsü, eş zamanlı fibrat/niasin/fusidik asit kullanımı gibi durumlar riski artırır.
Bu nedenle:
Pitavastatin başlanmadan önce risk faktörleri değerlendirilir,
Şiddetli kas ağrısı, güçsüzlük veya koyu renkli idrar gelişirse tedavi kesilir ve CK düzeyi değerlendirilir.
11.3. Hepatotoksisite
Pitavastatin, diğer statinlerde olduğu gibi:
Serum ALT/AST’de asemptomatik ve doz-bağımlı artışlara yol açabilir,
Nadir durumlarda klinik belirti veren karaciğer hasarı bildirilmiştir.
Tedavi öncesinde ve tedavi sırasında belirli aralıklarla karaciğer fonksiyon testlerinin izlenmesi; ALT/AST kalıcı olarak üst sınırın 3 katını aşarsa ilacın kesilmesi önerilir.
11.4. Diğer nadir ve ciddi reaksiyonlar
İmmün-aracılı nekrotizan miyopati (IMNM): Statinlere özgü otoimmün kas hastalığı tablosu pitavastatin ile de çok nadir bildirilmiştir.
İnterstisyel akciğer hastalığı: Uzun süreli statin tedavisi altında, dispne, kuru öksürük ve sistemik semptomlarla seyreden çok nadir olgular tarif edilmiştir.
Glukoz metabolizması: Sınıf etkisi olarak yeni başlangıçlı diyabet riskinde hafif artış statinlerle ilişkilendirilmekle birlikte, pitavastatin özelinde glisemik kontrol üzerine nötr veya daha elverişli bir profil bildiren çalışmalar vardır.
Genel olarak, pitavastatinin güvenlilik profili sınıf içi diğer statinlerle uyumlu olmakla beraber, CYP3A4 bağımlılığının düşük olması ve HDL-K üzerinde görece güçlü etkisi, belirli klinik bağlamlarda onu tercih edilir kılabilecek yönler olarak öne çıkar.
Keşif
Pitavastatin’in hikâyesi, yalnızca yeni bir statinin pazara çıkış öyküsü değil; Japon endüstriyel kimyasının, klinik kardiyolojinin ve ilaç regülasyon tarihinin birbirine sıkı sıkıya geçtiği, birkaç onyıla yayılan kolektif bir araştırma anlatısıdır.
1. Statinlerin “arka plan sahnesi”: Pitavastatin’e giden yol
1950’lerden itibaren kolesterol ve koroner kalp hastalığı arasındaki ilişki epidemiyolojik çalışmalarla giderek güçlendikçe, farmasötik endüstri için de kolesterol sentezini hedefleyen ilaçlar stratejik bir odak haline geldi. 1970’lerde Akira Endo ve çalışma arkadaşlarının mantar kaynaklı ilk HMG-CoA redüktaz inhibitörlerini (compactin, ardından lovastatin) tanımlaması, “statin çağını” başlatan dönüm noktasıydı.
Bu “doğal ürün kökenli” ilk statin dalgasını, 1980’ler ve 1990’larda tam sentetik statinlerin yükselişi izledi: fluvastatin, atorvastatin gibi moleküller, klasik farmakoforu korurken tamamen sentetik kimyasal iskeletler üzerine kurulmuştu. İşte pitavastatin, bu ikinci dalganın daha geç fakat rafine bir ürünü olarak, Japon ilaç Ar-Ge’sinin görece “geç ama güçlü” hamlelerinden biri şeklinde sahneye çıkacaktı.
2. Nissan Chemical ve Kowa’nın ortak programı: NK-104’ün doğuşu
1980’lerin sonuna gelindiğinde Japonya’da Nissan Chemical Industries (bugünkü Nissan Chemical Corporation) ve Kowa Company, kolesterol metabolizmasını hedef alan yeni nesil bir HMG-CoA redüktaz inhibitörü geliştirmek üzere ortak bir program başlattılar. Dünyada halihazırda çok sayıda statin varken Japon ekiplerin hedefi, özellikle üç noktada fark yaratabilecek bir molekül bulmaktı:
Yüksek potent ama orta dozlarda bile güçlü LDL-kolesterol düşüşü sağlayan,
CYP3A4’e çok az bağımlı, böylece çoklu ilaç kullanan hastalar için daha “uyumlu”,
HDL-kolesterol üzerine görece üstün bir etkisi olabilecek bir statin.
Bu programın kimyasal meyvesi, dahili kod adıyla NK-104 oldu. Molekül ilk aşamada, Nissan Chemical’in organik kimya ve medikal kimya birimlerinde çalışan bir ekip tarafından sentezlendi; bu ekibin bilimsel literatüre geçmiş görünen isimleri arasında, NK-104’ün sentez dizisini ayrıntılı biçimde yayımlayan K. Takahashi ve arkadaşları özellikle dikkat çeker. 1995 tarihli bu çalışma, kinolin çekirdeği ve siklopropil yan zincirle karakterize edilen özgün iskeletin adım adım sentezini tanımlayarak, daha sonra “pitavastatin” adını alacak molekülün medikal kimya “doğum kayıtlarından” birini oluşturdu.
Bugün klinik pratikte “keşif” tek bir bireye atfedilse de, pitavastatin gerçekte Nissan Chemical ve Kowa’nın çok sayıda medikal kimyager, farmakolog, toksikolog ve formülasyon uzmanından oluşan ortak Ar-Ge takımlarının ürünüdür. Sentez makaleleri, patent dosyaları ve erken faz farmakoloji çalışmaları birlikte okunduğunda, bireysel “mucit”ten çok, birkaç kuşak Japon araştırmacının sahip çıktığı kolektif bir proje görünümü ortaya çıkar.
Molekülün kimyasal iskeleti şekillendikten sonra sıradaki adım, onun gerçekten aranan “ideal” statine ne kadar benzediğini test etmekti. Bu dönemi, NK-104 kod adıyla yayımlanmış bir dizi farmakolojik ve farmakokinetik çalışma temsil eder.
3.1. Yapının farmakofora oturtulması
Takahashi ve ekibinin sentez çalışmasında ortaya konan iskelet, bilinen statin farmakoforunun temel öğelerini koruyarak, bunları yeni bir aromatik platforma yerleştiriyordu:
Enzimle etkileşen 3,5-dihidroksi heptenoik asit yan zinciri, mevalonata benzerliği ile HMG-CoA redüktazın aktif bölgesine kilitlenmeyi sağlıyordu.
Bu zincirin “taşıyıcısı” olarak seçilen 2-siklopropil-4-(4-florofenil) kinolin çekirdeği, molekülü güçlü biçimde lipofilikleştiriyor, karaciğer alımını kolaylaştırıyor ve enzim yüzeyindeki hidrofobik ceplere yerleşmesini mümkün kılıyordu.
Bu tasarım, klasik indol ve piridin türevli statinlerden ayrılan, daha “düz”, planarımsı, yoğun aromatik bir yüzey yaratıyordu; bu yüzey, redüktaz enziminin hidrokarbon cep bölgeleri ile daha sağlam π–π ve hidrofobik etkileşimler kurabilsin diye seçilmişti.
3.2. Erken farmakoloji: NK-104’ün güç sınavı
1990’ların sonuna gelindiğinde, NK-104 artık yalnızca bir sentetik hedef değil, deneysel hayvan modellerinde sistematik olarak test edilen bir adaydı.
K. Yamamoto ve arkadaşlarının yayınladığı çalışmalar, NK-104’ün çeşitli türlerde (rat, tavşan, köpek, maymun) HMG-CoA redüktazı son derece potent biçimde inhibe ettiğini ve LDL-benzeri partikülleri belirgin ölçüde düşürdüğünü göstermekteydi.
Hücre kültürü ve karaciğer perfüzyon deneyleri, sadece LDL reseptör ekspresyonunu artırmakla kalmayıp, VLDL ve IDL gibi trigliserit-zengin lipoproteinlerde de anlamlı azalmalar olduğunu ortaya koyuyordu.
Erken farmakokinetik veriler (intravenöz ve oral uygulamayı karşılaştıran hayvan ve insan çalışmaları), NK-104’ün hızlı emildiğini, karaciğerde yoğunlaştığını ve o dönemin diğer sentetik statinlerine göre nispeten uzun bir eliminasyon yarı ömrüne sahip olduğunu gösterdi.
Bu noktada devreye Kowa’nın klinik geliştirme ekipleri girdi: NK-104 için seçim çoktan yapılmıştı; molekül faz I–II klinik geliştirme programına doğru ilerliyordu.
4. Pitavastatin adının ortaya çıkışı ve patentlenme süreci
NK-104, klinik geliştirmede ilerledikçe ticari ve INN (International Nonproprietary Name) düzeyinde adlandırma süreci de başladı. Molekül, bu süreçte “itavastatin”, “nisvastatin” gibi geçici isimlerle de anıldı; nihayet “pitavastatin” adı altında tescil edildi.
Molekülün 1987 yılı civarında patent başvurusuna konu edildiği biliniyor; farklı yargı bölgelerinde, kinolin çekirdeğini ve 2-siklopropil-4-(4-florofenil) substitüsyonunu tanımlayan temel patentler bu yıllara tarihleniyor.
Takip eden yıllarda, pitavastatin kalsiyumun amorf ve kristalin formları, saflık sağlayan sentez yolları, tuz ve polimorf formları için çok sayıda ikinci nesil patent yayımlandı; bunların bir kısmı doğrudan Nissan Chemical ve Kowa’nın, bir kısmı da jenerik üretici firmaların adını taşıyor.
Burada “kaşifler” aslında patent belgelerinde “inventor” olarak listelenen, çoğu Japon ve Koreli kimyagerlerden oluşan çok sayıda isimdir; ancak bu listeler, farklı yargı bölgelerinde küçük değişikliklerle tekrarlandığından ve genellikle tek tek bireylerin bilimsel rollerini ayırt etmeye imkân vermediğinden, klinik literatürde genellikle “Nissan Chemical/Kowa araştırma ekipleri” ifadesi tercih edilir.
5. Klinik sahne: Japonya’dan dünyaya
5.1. Japon onayı ve erken klinik kullanım
2000’lerin başında yapılan pivotal çalışmalar tamamlandığında, pitavastatin için ilk büyük dönemeç, Japon düzenleyici otoritesi ve PMDA dosyalarında kayda geçti.
Japonya’da Livalo® tabletleri (1 ve 2 mg pitavastatin kalsiyum) için başvuruların ardından, Temmuz 2003’te hiperkolesterolemi ve ailesel hiperkolesterolemi tedavisi endikasyonlarıyla onay verildi.
Bu onayla birlikte Kowa, pitavastatini Japonya’da klinik pratikte yaygınlaştırmaya başladı; erken dönem gözlemsel veriler, özellikle HDL-kolesterol üzerindeki görece güçlü etki ve diyabetik hastalarda glisemik profile nötr etkiler açısından ilgi çekiciydi.
Bu dönemde literatürde adı sıkça geçen klinisyen araştırmacılar arasında K. Kajinami, J. Sasaki, Y. Saito gibi isimler öne çıktı; bunlar, pitavastatinin LDL-düşürücü profilini, HDL-arttırıcı kapasitesini ve pleiotropik etkilerini sistematik olarak raporlayan ilk klinik makalelerin başyazarlarıydı.
5.2. ABD ve Avrupa’ya açılış
Japonya’daki başarılı lansmandan sonra, pitavastatinin hedefi artık küresel pazardı:
2009 Ağustos’unda, ABD Gıda ve İlaç Dairesi (FDA), pitavastatin kalsiyumu Livalo® markası altında hiperkolesterolemi tedavisi için onayladı; bu, ABD piyasasında ruhsat alan yediinci statin anlamına geliyordu.
2010 yılında Birleşik Krallık’ta MHRA, Livazo® adı altında referans üye ülke (RMS) rolüyle pitavastatini değerlendirdi; bunu takiben birçok Avrupa ülkesinde ulusal onaylar alındı.
2017’de ise ABD’de farklı bir tuz formu olan pitavastatin magnezyum (Zypitamag®) için ek onay verildi; bu form, farmasötik açıdan eşdeğer kabul edilen ama fikrî mülkiyet açısından ayrı bir ürün olarak konumlandırıldı.
Bu genişleme sürecinde, pitavastatin artık yalnızca Japon Ar-Ge’sinin bir ürünü değil, uluslararası kılavuzların da içine yavaş yavaş giren bir lipid düşürücü ajan halini almaya başladı. Çalışmalarda tekrarlanan tema, orta-yüksek intensite LDL düşüşü ile birlikte belirgin HDL artışı ve CYP3A4’den bağımsız metabolizma kombinasyonuydu.
6. Pleiotropi ve mekanizma araştırmaları: Endotel, inflamasyon, anjiyogenez
Pitavastatin klinik olarak yerini buldukça, araştırmacıların ilgisi molekülün kolesterol dışı etkilerine, yani “pleiotropik” özelliklerine doğru kaymaya başladı.
2010’lu yılların başında J. Davignon ve diğer araştırmacılar, pitavastatinin endotel fonksiyonunu nasıl iyileştirdiğini, nitrik oksit sentezini artırdığını, vasküler inflamasyonu ve oksidatif stresi azalttığını derleyen çalışmalar yayımladılar.
Kikuchi ve ekibi, pitavastatinin endotel hücrelerinde Notch1 sinyal yolaklarını ve γ-sekretaz aktivitesini modüle ederek anjiyogenezi artırabildiğini; üstelik bunun VEGF bağımsız gerçekleştiğini gösteren mekanistik veriler sundu.
Bu mekanistik çalışmalar, pitavastatinin klinik düzeyde gözlenen bazı “ekstra” etkilerini (örneğin, vasküler yeniden şekillenme süreçleri, plak stabilitesi, inflamatuvar biyobelirteçlerde azalma) biyokimyasal düzeyde anlamlandırmaya yardım etti.
7. Yeni sahne: HIV, epigenetik yaşlanma ve REPRIEVE çağı
Pitavastatinin tarihindeki en dramatik ikinci kırılma noktası, 2020’lerin başında HIV ile yaşayan bireylerde yürütülen büyük ölçekli kardiyovasküler sonuç çalışmalarıyla geldi.
REPRIEVE (Randomized Trial to Prevent Vascular Events in HIV) çalışması, düşük–orta aterosklerotik riskli, antiretroviral tedavi altında HIV ile yaşayan binlerce bireyi, pitavastatin 4 mg/gün ve plasebo kollarına randomize etti. 2023’te yayımlanan sonuçlar, pitavastatin kolunda majör advers kardiyovasküler olaylarda belirgin azalma göstererek, statinin bu hasta grubunda primer koruma amacıyla standartlara girmesinin önünü açtı.
REPRIEVE verilerini takiben, 2025’te yayımlanan analizler, pitavastatinin bu popülasyonda kardiyovasküler olayları azaltırken, AIDS-dışı malignite riskini anlamlı biçimde düşürmediğini; buna rağmen genel klinik yararın güçlü olduğunu ortaya koydu.
Aynı kohorttan türeyen yan çalışmalar, pitavastatinin epigenetik yaşlanma biyobelirteçleri üzerindeki olası etkilerini araştırmaya başladı; erken sonuçlar, statin tedavisinin HIV ile yaşayan bireylerde hızlanmış biyolojik yaşlanma dinamiklerini kısmen modüle edebileceği hipotezini gündeme getirdi.
Bu noktada pitavastatin, artık yalnızca “bir başka statin” değil, HIV alanında primer kardiyovasküler koruma için özel olarak seçilmiş bir molekül kimliğine kavuşmuş durumdadır; kılavuzlar ve uzman gruplar, HIV ile yaşayan bireylerde statin seçiminde pitavastatini belirgin biçimde öne çıkaran öneriler yayımlamaktadır.
8. Karaciğer ve metabolizma cephesi: NAFLD/MASLD, NASH ve ötesi
Pitavastatin’in hikâyesi yalnızca damar duvarıyla bitmiyor; karaciğer ve metabolik hastalıklar ekseninde de yeni bölümler yazılıyor.
2010’ların başında H. Hyogo ve arkadaşlarının yürüttüğü, biyopsi ile kanıtlanmış NASH hastalarında 2 mg/gün pitavastatin tedavisini inceleyen çalışma, hem lipid profilinde iyileşme hem de bazı olgularda histolojik düzelme bulguları bildirdi; bu, daha büyük NASH/NAFLD çalışmalarına ilham veren erken bir sinyal olarak kabul edildi.
Sonraki yıllarda klinik kayıtlar ve deneysel modeller, pitavastatinin karaciğer yağlanması, inflamasyonu ve fibrozisi üzerindeki etkilerini ayrıntılandırdı; 2020’lerin ortasında yayımlanan hayvan çalışmaları, yüksek yağ–yüksek fruktoz diyetle indüklenen NAFLD modellerinde steatoz ve fibrozun anlamlı ölçüde gerilediğini göstererek bu ilgi alanını daha da güçlendirdi.
Öte yandan, randomize kontrollü insan çalışmalarında pitavastatinin insülin duyarlılığı üzerindeki etkisinin büyük ölçüde nötr olduğu, buna karşın karaciğer yağ içeriği üzerinde hafif ama klinik olarak ilginç olabilecek düzeyde azalma sağlayabildiği gösterildi.
Bu veriler, karaciğer hastalıklarında statin kullanımına dair eskiden yaygın olan “çekingen yaklaşımın” yerini, daha güvenli ve proaktif bir kullanıma bırakmasında rol oynadı; pitavastatin bu tartışmanın merkezinde yer alan moleküllerden biri oldu.
9. Onkoloji sahnesi: Üçlü negatif meme kanseri ve metabolik hedefleme
Pitavastatinin hikâyesinin en yeni ve belki de en beklenmedik bölümü, kanser biyolojisi ile ilgili. Kolesterol biyosentezi, tümör hücrelerinin proliferasyonu ve hayatta kalmasında kritik bir rol oynadığından, statinler uzun süredir potansiyel antikanser ajanlar olarak tartışılıyor; ancak bu alan, yeni nesil tarama teknolojileriyle başka bir boyut kazandı.
2024’te Alissandra L. Hillis ve geniş bir araştırma konsorsiyumu, üçlü negatif meme kanserinde (TNBC) yapılan CRISPR taramalarının ardından, kolesterol biyosentezi yolunu metabolik bir zayıf nokta olarak tanımladı. Bu çalışmada, pitavastatin gibi güçlü HMG-CoA redüktaz inhibitörlerinin, AKT inhibitörleri (ör. capivasertib/AZD5363) ile kombine edildiğinde TNBC hücrelerinde belirgin sentetik öldürücülük (synthetic lethality) oluşturduğu gösterildi.
Bu bulgular, kolesterol biyosentezi–PI3K/AKT ekseni kesişiminde yeni bir antikanser stratejinin kapısını aralıyor; pitavastatin burada, zaten onaylı ve güvenlik profili iyi bilinen bir ilaç olarak, “yeniden konumlandırma” (drug repurposing) açısından avantajlı konumda.
Onkoloji alanındaki bu çalışmalar henüz erken fazda ve klinik rutine taşınmış değil; fakat pitavastatinin evrimsel hikâyesine, saf lipid düşürücü ajanlıktan tümör metabolizmasını hedefleyen bir ortak oyuncuya dönüşme potansiyelini ekliyor.
10. Güncel değerlendirmeler: 2020’lerin ortasında pitavastatin
Bugün, 2020’lerin ortasına gelindiğinde, pitavastatin hakkında yayımlanan sistematik derlemeler ve uzman görüş yazıları, molekülü birkaç temel eksende konumlandırıyor:
LDL-kolesterolü orta–yüksek intensite aralığında etkin biçimde düşüren,
HDL-kolesterolü görece güçlü artıran,
CYP3A4’ten bağımsız metabolizması sayesinde polifarmasi ortamında avantajlı,
Endotel fonksiyonunu, inflamasyonu ve oksidatif stresi olumlu etkileyen pleiotropik bir statin.
Özellikle HIV ile yaşayan bireyler, metabolik sendrom ve NAFLD/MASLD spektrumu ve onko-metabolik araştırmalar, pitavastatinin hikâyesinin yeni bölümlerinin yazıldığı başlıca sahneler olarak öne çıkıyor. Japonya’da birkaç kimyagerin kinolinli bir statin fikriyle başlayan bu yolculuk, bugün hem klasik kardiyovasküler risk azaltımının hem de karmaşık metabolik–inflamatuvar ve onkolojik süreçlerin kesişim noktasında, hâlâ genişlemeye devam eden bir araştırma alanına dönüşmüş durumda.
İleri Okuma
Yagi, S. (2008). Pitavastatin, an HMG-CoA Reductase Inhibitor, Exerts Pleiotropic Actions on Cardiovascular Remodeling. Circulation Research, 102(6), 689–699. doi:10.1161/CIRCRESAHA.107.163493
U.S. Food and Drug Administration (2009). Livalo (pitavastatin) Tablets, NDA 022363 – Approval Package. Silver Spring, MD: FDA.
Saito, Y. (2009). Critical appraisal of the role of pitavastatin in treating dyslipidemias and achieving lipid goals. Vascular Health and Risk Management, 5, 921–936.
Sasaki, J. (2010). Pitavastatin approved for treatment of primary hypercholesterolemia and combined dyslipidemia. International Journal of General Medicine, 3, 349–357.
Watson, K. E. (2010). Pitavastatin: The Newest HMG-CoA Reductase Inhibitor. Reviews in Cardiovascular Medicine, 11(1), 38–46. doi:10.3909/ricm0535
European Medicines Agency / MHRA (2010–2011). Livazo/Alipza/Vezepra/Pitavastatin Public Assessment Report and SmPC. UK/H/1555-8/001-3/DC.
Iwata, H., et al. (2012). Safety and efficacy of pitavastatin in patients with dyslipidemia: Results of a pooled analysis of clinical trial data. Clinical Drug Investigation, 32(3), 183–195.
McKenney, J. M., & Koren, M. J. (2012). Pitavastatin: An evaluation of its pharmacokinetics, clinical efficacy and tolerability. Expert Opinion on Drug Metabolism & Toxicology, 8(7), 887–902.
Ballantyne, C. M., et al. (2013). Efficacy and safety of pitavastatin in patients with primary hyperlipidemia or mixed dyslipidemia. The American Journal of Cardiology, 112(2), 191–199.
Koh, K. K., et al. (2013). Comparative effects of pitavastatin versus atorvastatin on lipoprotein metabolism and inflammatory biomarkers in patients with hypercholesterolemia. Atherosclerosis, 230(2), 273–279.
Vallejo-Vaz, A. J., et al. (2017). Efficacy and safety of pitavastatin in individuals with atherosclerotic cardiovascular disease and diabetes: A systematic review and meta-analysis. Cardiovascular Diabetology, 16(1), 1–14.
Kowa Pharmaceuticals (2019). About Pitavastatin – QFACTS Medical Information Brochure.
National Institutes of Health (2021). LiverTox: Clinical and Research Information on Drug-Induced Liver Injury – Pitavastatin. NCBI Bookshelf.
Ramadan, A., et al. (2022). Mini-Review on the Efficacy and Safety of Pitavastatin. Journal of Clinical Medicine, 11(16), 4800. doi:10.3390/jcm11164800
Bhatti, H., & Jneid, H. (2023). Pitavastatin. In StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing.
Qeios (2024). Pitavastatin: A Comprehensive Overview of its Mechanisms, Pharmacokinetics, Pharmacodynamics, and Adverse Effects. Qeios ID: FW9ZT3.
U.S. Food and Drug Administration (2024). LIVALO (pitavastatin) – Prescribing Information (Label 022363s022).
Shi, Z., et al. (2025). Personalized statin therapy: Targeting metabolic processes and hepatic uptake transporters. Heliyon, 11, eXXXXX.
Kowa Pharmaceuticals Europe (erişim 2025). Livazo® (pitavastatin) – Summary of Product Characteristics, 1 mg/2 mg/4 mg film-coated tablets (konsolide sürüm).
PubChem. Pitavastatin – Chemical identifiers and compound summary.
Clabin® (salisilik asit + laktik asit + resorsinol içeren siğil ve nasır solüsyonu)
1. Giriş
Clabin®, keratolitik bir topikal solüsyon olarak geliştirilen ve ağırlıklı olarak siğil (verruka) ve nasırların tedavisinde kullanılan klasik bir eczacılık preparatıdır. Formülasyonu, keratinize dokuyu parçalayan ve yumuşatan üç temel etkin maddeye dayanır: salisilik asit, laktik asit ve resorsinol. Bu üçlü, kolodyon benzeri bir taşıyıcı sistem içinde çözünmüş hâlde deriye uygulanır ve özellikle el, ayak ve parmaklarda görülen verrukalar ile mekanik basınca bağlı nasırların kademeli olarak uzaklaştırılmasını amaçlar.
Clabin®’in ilk pazarlama izninin 1962 yılında verilmiş olması, onu modern dermatolojik OTC (over-the-counter / reçetesiz) keratolitik tedavilerin erken örneklerinden biri hâline getirir. 2001 yılında Amerika Birleşik Devletleri’nde piyasaya sürülmesini takip eden dönemde, özellikle hafif ve orta şiddette verrukalar için reçetesiz başvuru ürünlerinden biri olarak yaygınlaşmıştır. Üretici firma olarak Chattem, Inc. belirtilir.
2. Etimoloji: İsimler ve kavramların kökeni
2.1. “Clabin” adı
“Clabin” ticari adı muhtemelen doğrudan klasik bir dil köküne değil, marka yaratımı yoluyla türetilmiş bir kelimeye dayanır. Ancak fonetik yapısı, Latince “clavus” (nasır, çivi gibi batıcı sert oluşum; tıpta nasır için de kullanılır) terimini çağrıştırır. Nasır ve siğil tedavisine yönelik bir preparat için “clav-” köküne sesçe yaklaşan bir isim seçilmesi, muhtemelen bilinçli bir ticari ve semantik tercih olarak değerlendirilebilir.
2.2. “Siğil” ve “verruka”
Türkçe “siğil” sözcüğü, köken olarak Eski Türkçeye uzanır ve deride “kabarık, pürtüklü oluşum” anlam alanında yer alır. Modern tıpta ise Latince “verruca” terimi hâkimdir; bu sözcüğün, yine “pürtüklü, kaba yüzey” anlamıyla ilişkili eski İtalik köklerden geldiği kabul edilir. Klinik terminolojide “verruka vulgaris” yaygın, sıradan siğil; “verruka plantaris” ise ayak tabanındaki plantar siğil için kullanılır.
2.3. Etkin madde adlarının etimolojisi
Salisilik asit: İsmini, söğüt anlamına gelen Latince Salix’ten alır. Tarihsel olarak salisilat türevleri, söğüt kabuğundan elde edilen analjezik ve antipiretik maddeler üzerinden tanımlanmıştır.
Laktik asit: Latince lac, lactis (süt) sözcüğüne dayanır. Laktik asit ilk olarak ekşi sütten izole edildiği için bu şekilde adlandırılmıştır.
Resorsinol: Kimyasal adlandırmada “rezorsinol” veya “resorsinol” şeklinde geçen bu bileşik, başlangıçta “rezin” (lat. resina) türevleriyle, ayrıca “orcinol” adı verilen başka bir fenolik bileşikle olan yapısal benzerliği üzerinden adlandırılmıştır. İsim, organik kimyada fenolik bileşikler için kullanılan tarihsel adlandırma geleneklerinin bir ürünüdür.
Bu etimolojik arka plan, Clabin® içeriğindeki bileşenlerin, bitkisel kaynaklı tıbbi gözlemlerden rafine edilmiş kimyasal moleküllere uzanan tarihsel bir çizginin ürünü olduğunu gösterir.
3. Tarihçe ve düzenleyici arka plan
Clabin®’in ilk pazarlama izni 1962 yılında verilmiş, böylece keratolitik kombinasyon preparatlarının eczane raflarında standart hâle gelmeye başladığı dönemi temsil eden ürünlerden biri olmuştur. O yıllarda siğil tedavisi; kriyoterapi gibi ofis içi girişimsel işlemlerden daha sınırlı erişilebilir durumdaydı ve uzun süreli topikal keratolitik tedaviler, birçok hasta için ilk basamak seçenek konumundaydı.
2001 yılında ABD’de piyasaya sürülmesi, ürünü küresel OTC pazarına daha güçlü bir şekilde taşımış; özellikle eczane zincirlerinde “wart remover” kategorisinde salisilik asitli ürünler arasında yer almasına olanak sağlamıştır. Bu tarih, ABD’de OTC siğil tedavilerinin yoğunlaştığı bir dönemle çakışır; jel, sıvı, ped ve bant formundaki salisilik asit ürünleri arasında Clabin® tarzı kolodyon-tipi preparatlar da yer almıştır.
4. Farmasötik formülasyon ve bileşenler
Clabin® solüsyonu, etkin maddelerin keratinize doku üzerine odaklı ve kontrollü şekilde bırakılmasını sağlayan, uçucu solventler ve film oluşturan ajanlardan oluşan bir taşıyıcı faz içerisinde formüle edilmiştir.
4.1. Etkin maddeler
Salisilik asit
Temel rolü keratolitik etkidir: Stratum korneumdaki keratin ağını kimyasal olarak çözerek, hiperkeratotik siğil dokusunu yavaş yavaş parçalar.
Desmosomlar ve hücreler arası bağları zayıflatarak, korneositlerin daha kolay dökülmesini sağlar.
Daha yüksek konsantrasyonlarda keratolitik, daha düşük konsantrasyonlarda keratoplastik (yani keratin tabakasının yeniden düzenlenmesini teşvik eden) etki gösterebilir.
Laktik asit
Literatürde hem hafif keratolitik hem de humektan (nem çekici) özellikleriyle tanımlanır.
Epidermise su bağlama kapasitesini artırır, siğil dokusunu yumuşatır ve böylece salisilik asidin daha derin ve homojen nüfuz etmesine yardım eder.
Fenolik yapıda, hafif antiseptik, hafif keratolitik ve zayıf dezenfektan etkileri olan bir bileşiktir.
Siğil yüzeyinde bakteri kolonizasyonunu sınırlamaya yardımcı olabilir ve keratinizasyonu modüle ederek salisilik asidin keratolitik etkinliğini tamamlar.
4.2. Yardımcı maddeler (eksipiyanlar)
Eter: Uçucu bir organik çözücü olarak, etkin maddelerin çözünürlüğünü artırır. Uygulama sonrası hızla buharlaşarak etkin maddelerin daha yoğun halde siğil yüzeyinde kalmasını sağlar.
Piroksilin (nitroselüloz): Kolodyonun temel film oluşturucu bileşenidir. Uygulama sonrasında deride ince, yapışkan bir film tabakası oluşturarak preparatın lokalize kalmasına, mekanik koruma sağlamasına ve etkin maddelerin kontrollü salınımına yardım eder.
Etil asetat: Çözücü ve seyreltici olarak kullanılır; uçucu özelliği sayesinde uygulama sonrası hızla uzaklaşır, film tabakasının kısa sürede sabitlenmesini destekler.
Bu bileşenler birlikte, siğil ya da nasır üzerine sürüldükten sonra hızla uçup geride etkin maddelerle zenginleşmiş ince bir film bırakan, klasik bir “kolodyon-tipi keratolitik solüsyon” oluştururlar.
5. Patogenez ve etki mekanizmasının biyolojik temeli
5.1. Verrukaların biyolojisi ve HPV
Çoğu siğil, insan papilloma virüsünün (HPV) belirli tipleriyle oluşur. HPV, epidermisin bazal tabakasındaki keratinositlere yerleşir ve bu hücrelerin proliferasyonunu artırarak karakteristik hiperkeratotik, pürtüklü lezyonları oluşturur.
Evrimsel açıdan bakıldığında HPV, konak keratinositlerinin doğal farklılaşma programını “hackleyen” bir virüstür. Virüs, keratinositlerin bölünme ve farklılaşma döngüsünü kendi replikasyonuna uygun şekilde yeniden düzenler; bu da kalınlaşmış, keratin yüklü bir doku alanı yaratır. Bu alan, konakta kozmetik ve fonksiyonel sorunlara yol açsa da virüs için nispeten güvenli bir çoğalma nişidir.
5.2. Keratin tabakasına yönelik kimyasal “saldırı”
Clabin®’deki etkin maddelerin tüm stratejisi, HPV’nin yerleştiği hiperkeratotik dokunun aşamalı olarak parçalanmasına dayanır:
Resorsinol, hem keratolitik süreci destekler hem de lokal antiseptik etkiyle ikincil enfeksiyon riskini azaltmaya yardımcı olur.
Bu kombinasyon, siğil dokusunun katman katman soyulmasına yol açarken, virüsün yaşadığı ve çoğaldığı hücre popülasyonunu da ortadan kaldırmayı amaçlar.
5.3. Evrimsel açı: Neden keratolitik tedaviler işe yarar?
HPV, keratin sentezini ve keratinosit proliferasyonunu güçlendirerek kendine kalın bir “keratin zırhı” yaratır. Evrimsel süreçte bu strateji, virüsün dış ortamdan ve immün yanıtın bazı bileşenlerinden korunmasına yardımcı olur. Ancak bu zırh, klinisyen ve hasta açısından bir zayıf nokta hâline getirilebilir: Keratini hedef alan kimyasal ajanlar (salisilik asit, laktik asit, üre vb.) bu kalın tabakayı kontrollü şekilde çözdüklerinde, virüsün nişi fiziksel olarak ortadan kalkar.
Clabin® gibi keratolitik kombinasyonlar, aslında HPV’nin evrimsel stratejisinin bir yan ürününü—kalın keratin tabakasını—tedavinin hedefi hâline getirir. Bu bakış açısıyla preparat, doğrudan antiviral bir ilaç olmaktan çok, virüsün inşa ettiği “mikro-ekosistemi” çökerten bir kimyasal araç olarak görülebilir.
6. Farmakokinetik yönler
Clabin® topikal olarak, sınırlı bir alana uygulanır. Normal kullanım koşullarında:
Etkin maddelerin sistemik emilimi düşüktür.
Saçlı deriden, mukozal yüzeylerden veya geniş erozyon alanlarından uygulama yapılmadığı sürece, serum salisilat düzeylerinde klinik anlamlı artış beklenmez.
Nitroselüloz film tabakası, ilacın sistemik dolaşıma değil, lokal epidermal katmanlara yönelmesini destekler.
Bununla birlikte, özellikle çocuklarda, geniş alanlara, çatlak veya inflamasyonlu deriye uygulama durumunda teorik olarak sistemik salisilat maruziyeti artabilir. Bu nedenle ürün, kullanım bilgilerinde genellikle belirli anatomik bölgelerle sınırlama, küçük alanlarla yetinme ve önerilen sürenin aşılmaması uyarılarıyla birlikte sunulur.
7. Endikasyonlar
Clabin® solüsyonunun temel klinik kullanım alanları şunlardır:
Siğiller (verrukalar)
Verruka vulgaris (el ve parmaklarda klasik siğiller)
Bazı plantar siğiller (ayak tabanında, basınç alanlarında; uygun vakalarda)
Özellikle ayak parmakları üzerinde veya tabanda yerleşen, “çivi gibi batan” nasır oluşumları
Formülasyon, hiperkeratotik lezyonları gidermeyi, nasır ve siğil yüzeyini yumuşatmayı ve zamanla bu dokunun mekanik veya spontan olarak düşmesini hedefler.
8. Uygulama şekli, doz ve tedavi süresi
Clabin® klasik olarak, küçük bir aplikatör veya pamuklu çubuk yardımıyla lokal olarak uygulanır. Kullanım şeması genel olarak şu şekilde özetlenebilir:
Etkilenen bölge ılık suyla birkaç dakika yumuşatılır, ardından dikkatlice kurulanır.
Gerekiyorsa nasır veya siğil yüzeyindeki kalın keratin tabakası, dikkatli bir şekilde törpülenir (sağlam deriye zarar vermeden).
Clabin® solüsyonu, pamuklu çubuğun ucuyla yalnızca siğil veya nasır üzerine ince bir tabaka hâlinde sürülür; çevredeki sağlıklı deriye temas ettirilmemelidir.
Solventler uçup nitroselüloz filmi sabitlenene kadar bölge birkaç dakika açık bırakılır.
Ürün genellikle günde bir kez uygulanır.
Tedavi süresi en fazla 12 haftaya kadar uzatılabilir.
Eğer 12 haftalık düzenli kullanıma rağmen siğil kaybolmaz veya belirgin bir küçülme göstermezse, olası alternatif tanılar, yanlış uygulama tekniği veya daha dirençli HPV tipleri açısından bir dermatolog tarafından tekrar değerlendirme önerilir.
9. Kontrendikasyonlar ve dikkat edilmesi gereken durumlar
Clabin® kullanımı için bildirilen başlıca kontrendikasyonlar şunlardır:
Gebelik ve emzirme: Ürün gebelikte ve emzirme döneminde önerilmemektedir. Salisilik asidin sınırlı da olsa sistemik emilimi, özellikle geniş alan uygulamalarında teorik risk oluşturabilir.
Açık yaralar ve fissürler: Erozyonlu, ülserli veya derin çatlaklı alanlarda kullanımı uygun değildir; bu durum sistemik emilim riskini artırır ve yoğun ağrıya yol açabilir.
Salisilik asit, laktik asit veya resorsinole karşı bilinen aşırı duyarlılık: Anamnezinde bu bileşenlere karşı alerjik kontakt dermatit veya ciddi irritan reaksiyon öyküsü olan hastalarda kullanılmamalıdır.
Ek dikkat gerektiren durumlar:
Diyabetik ayak sendromu veya periferik arter hastalığı olan hastalarda, özellikle ayaklarda nasır ve siğil tedavisi için self-medikasyon (kendi kendine tedavi) yerine mutlaka hekim danışmanlığı önerilir.
Çocuklarda daha küçük alanlara, daha kısa süreli uygulama tercih edilmeli; yetişkinler için tasarlanan şemalar otomatik olarak uygulanmamalıdır.
10. Yan etkiler ve istenmeyen reaksiyonlar
Clabin®, önerilen şekilde kullanıldığında çoğu birey tarafından iyi tolere edilen bir preparattır. Bununla birlikte, lokal olarak ortaya çıkabilen yan etkiler gözlenebilir:
Hafif cilt tahrişi
Uygulama bölgesinde yanma veya batma hissi
Eritem (kızarıklık)
Kaşıntı
Hafif soyulma veya pullanma
Bu reaksiyonlar, genellikle keratolitik etkinin beklendik bir uzantısıdır ve lezyonla sınırlı, tolere edilebilir düzeyde seyreder. Ancak:
Belirgin ağrı
Yaygın kızarıklık
Kabarcık oluşumu
Çevre sağlıklı deride belirgin tahriş
gibi bulgular durumda ürünün kullanımı derhâl kesilmeli ve hekim değerlendirmesi istenmelidir.
Nadir de olsa resorsinol ve salisilik asit, duyarlı kişilerde kontakt dermatite yol açabilir. Çok geniş alanlara uzun süreli ve yoğun uygulama teorik olarak salisilat toksisitesi riskini artırabilir; ancak Clabin® gibi lokal ve sınırlı alan preparatlarında bu risk normal koşullarda oldukça düşüktür.
11. İlaç etkileşimleri ve özel hasta grupları
Topikal, sınırlı alan uygulaması nedeniyle klasik anlamda sistemik ilaç etkileşimi beklenmez. Yine de:
Aynı bölgeye başka iritan veya eksfolyan ürünlerin (örneğin güçlü asitli peeling ajanları, retinoidler) eşzamanlı uygulanması, irritasyon ve kimyasal yanık riskini artırabilir.
İmmünsupresif tedavi alan ve yaygın HPV enfeksiyonu bulunan hastalarda, yalnızca keratolitik tedavi yetersiz kalabilir; bu hasta grubunda dermatolog kontrolü esastır.
Çocuklar, yaşlılar, diyabetliler, periferik damar hastalığı olanlar ve nöropatisi bulunanlar (özellikle ayak tabanı uyguları için) özel dikkat gerektiren gruplardır.
12. Diğer tedavi seçenekleriyle karşılaştırma
Clabin® gibi keratolitik kombinasyonlar, verruka tedavi algoritmasında genellikle şu tedavi yöntemleriyle karşılaştırılır:
Kriyoterapi (sıvı azot): Daha hızlı, klinik ortam gerektiren, bazen daha ağrılı olabilen bir yöntemdir. Özellikle tek veya az sayıdaki siğil için tercih edilebilir.
Yalnız salisilik asit içeren bant, ped veya solüsyonlar: Daha basit formülasyonlar olup benzer etki mekanizmasına sahiptir; Clabin®’den temel farkları, laktik asit ve resorsinol gibi ek yardımcı keratolitik ve antiseptik bileşenleri içermemeleridir.
İmikimod, podofilotoksin, kantaridin vb.: Daha spesifik veya güçlü etki mekanizmalarına sahip, genellikle hekim reçetesi ile kullanılan ajanlardır; bazıları immünomodülatör ya da sitotoksik etki gösterir.
Lazer, küretaj, elektrokoter: Daha invazif, lokal anestezi gerektirebilen yöntemlerdir ve genellikle dirençli ya da kozmetik açıdan sorunlu lezyonlarda kullanılır.
Clabin® ve benzeri keratolitik OTC ürünler, özellikle küçük, sınırlı sayıda ve komplikasyonsuz verrukalar için, düşük maliyetli ve evde uygulanabilir ilk basamak seçenekler arasında yer alır.
13. Klinik kullanım perspektifi
Clabin® kullanımında klinik açıdan önemli olan, hastaya doğru uygulama tekniğinin ve sürekliliğin vurgulanmasıdır. Verruka tedavisi çoğu zaman sabır gerektirir; keratolitik ajanlarla tedavi haftalar boyunca düzenli kullanım gerektirir ve erken kesilen uygulamalar çoğu zaman “etkisiz kaldı” şeklinde yanlış algılanabilir.
Öte yandan, 12 haftalık bir kullanım süresine rağmen kayda değer bir iyileşme yoksa, altta yatan tanının yeniden gözden geçirilmesi (örneğin, siğil zannedilen lezyonun başka bir dermatoz veya nadiren bir neoplazi olması) önemlidir. Bu nedenle uzun süreli ve sonuçsuz OTC kullanımlarında dermatolojik muayene geciktirilmemelidir.
14. Alternatif siğil ilaçları
Clabin®’e ek olarak piyasada çok sayıda alternatif siğil ilacı bulunur. Bunlar kabaca birkaç gruba ayrılabilir:
Sadece salisilik asit içeren solüsyon, jel, bant ve pedler
Üre, laktik asit veya triklorasetik asit içeren keratolitik ürünler
Hekim reçetesiyle verilen topikal immünomodülatörler (örneğin imikimod)
Fitoterapötik veya “doğal” içerikli, etkinliği daha sınırlı ve heterojen ürünler
Bu seçenekler arasında seçim; lezyonun lokalizasyonu, sayısı, hastanın ağrı toleransı, altta yatan komorbiditeler ve hastanın tedaviden beklentilerine göre yapılmalıdır. Clabin® bu spektrum içinde, klasik ve iyi tanımlanmış bir keratolitik kombinasyon preparatı olarak, özellikle evde kendi kendine uygulanabilen, kontrollü fakat görece yavaş bir tedavi yöntemi sunar.
Keşif
1. Başlangıç: 19. Yüzyılın Kimyasal Devrimleri
Clabin®’in kökeni, ürünün kendisinden çok önce, 19. yüzyıl kimyasının derin dönüşümleriyle başlar. Bu dönem, organik kimyanın henüz bir keşif alanı olduğu, laboratuvarların yeni moleküllerle dolup taştığı yıllardır.
1.1. Salisilik asidin sentezi ve Hermann Kolbe’nin laboratuvarı
Hikâye, 1850’lerde Alman kimyager Hermann Kolbe ile başlar. Kolbe, o dönemde büyük bir bilimsel değişim yaratarak, bitkilerden elde edilen bir doğal ürün olan salisilik asidi tamamen laboratuvar ortamında sentezlemeyi başarır. Söğüt kabuğundan elde edilen bu antik ağrı kesicinin kimyasal olarak üretilmesi, yalnız farmakolojide değil, modern dermatolojik tedavilerin geleceğinde de belirleyici olur.
1.2. Laktik asidin tanımlanması: Scheele’den Wöhler’e uzanan yol
Clabin®’in ikinci temel bileşeni olan laktik asit ise daha erken tarihlerde İsveçli kimyager Carl Wilhelm Scheele tarafından ilk kez tanımlanmış, ardından 19. yüzyılın başlarında Friedrich Wöhler tarafından biyokimyasal işlevleri sistematik şekilde araştırılmıştır. Sütün ekşimesiyle ilişkili bu asidin keratini yumuşatma etkisi henüz bilinmese de, ileride Clabin®’in etkinliğinin kilit bileşenlerinden biri olacağı açıktır.
1.3. Resorsinolün keşfi: Heinrich Hlasiwetz ve fenolik bileşiklerin doğuşu
Clabin®’in üçüncü ayağı olan resorsinol ise 1860’larda Avusturyalı kimyager Heinrich Hlasiwetz tarafından karakterize edilmiştir. Resorsinol, antiseptik gücü ve keratinle etkileşim kurabilme kapasitesiyle erken dermatolojik formüllerde hızla yer edinmiştir.
Bu üç bileşenin bağımsız keşfi, bir araya geldiklerinde oluşturacakları terapötik etkiden henüz habersizdi; fakat Clabin®’in geleceği bu kimyasal buluşlar üzerine kurulacaktı.
yüzyılın sonlarında nitroselülozun (pyroksilin) keşfi, yalnızca sinema ve endüstriyel kaplamaları değil, tıbbi formülasyonları da dönüştürür. Piroksilin çözüldüğünde, uygulandığı yüzeyde ince ve dayanıklı bir film bırakır. Kimyagerler kısa sürede bu özelliğin ilaç taşıyıcı sistemlerde kullanılabileceğini fark eder.
Eczacılar, salisilik asit, laktik asit ve resorsin gibi keratini yumuşatan maddeleri kolodyon matrisi içine entegre etmeye başlar. Bu formülasyonlar, dönemin tıp kitaplarında “verruka ve kallusları eritmeye yarayan preparatlar” olarak tanımlanır.
Bu dönem, Clabin®’in doğrudan atası sayılabilecek ürünlerin ilk defa ortaya çıktığı bir geçiş dönemidir.
3. Bilimsel Standardizasyon: 20. Yüzyılın İlk Yarısı
3.1. Eczanelerde hazırlanan karışımlardan endüstriyel formüllere geçiş
1930–1950 yılları arasında, dermatolojik ajanların güvenirliği ve bileşim oranları üzerine ciddi standartlaştırma çalışmaları başlar. Bu dönemde, üçlü kombinasyon (salisilik asit + laktik asit + resorsinol) giderek daha tutarlı hâle gelir.
3.2. Dünya çapında benzer ürünlerin yaygınlaşması
Bu yıllarda özellikle Avrupa’da ve ABD’de, eczanelerde satılan çok sayıda siğil ve nasır giderici formül ortaya çıkar. Ürünler ad değiştirir, formülasyonlar geliştirir, ancak kombinasyon hep benzerdir: keratini çözen salisilik asit, deriyi yumuşatan laktik asit, dokuyu stabilize edip dezenfekte eden resorsinol.
Bu zincir, Clabin®’in ortaya çıkması için gerekli tüm teknik birikimi hazırlamıştır.
4. Clabin® Markasının Doğuşu (1962)
4.1. İlk ruhsatlandırma
1962 yılında, modern eczacılık endüstrisinin olanaklarıyla stabilize edilmiş, kontrollü oranlara sahip bir keratolitik kolodyon preparatı, Clabin® adıyla ilk kez pazarlama izni alır. Ticari marka ismi, Latince clavus (nasır, çivi gibi sert kütle) sözcüğünü çağrıştırır.
4.2. Chattem, Inc. ile uluslararasılaşma
Sonraki yıllarda üretim ve distribüsyon Chattem, Inc. tarafından devralınır. Firma, dermatolojik OTC ürünlerindeki uzmanlığıyla formülün raf ömrünü artırır, uygulama kolaylığını geliştirir ve Clabin®’i uluslararası pazarlara yayar.
5. ABD Pazarına Giriş ve Yaygınlaşma (2001)
2001 yılı, Clabin® için önemli bir dönüm noktasıdır. Bu tarihte ürün, ABD’de bir OTC siğil tedavisi olarak satışa sunulur. Aynı dönemde Amerikan piyasasında siğil tedavileri hızla yaygınlaşmakta; evde uygulanan keratolitik ve kriyoterapi benzeri ürünlere ilgi artmaktadır. Clabin® bu ortamda güvenilir, klasik ve etkin bir seçenek olarak yerini alır.
6. Modern Araştırmalar: HPV Biyolojisi ve Keratolizme Yeni Bakış (2000–Günümüz)
6.1. HPV’nin hücresel ekolojisinin çözülmesi
Son yirmi yılda HPV biyolojisi derinlemesine araştırılmıştır. Virüsün keratinositleri manipüle ederek kendine “koruyucu bir keratin kalesi” oluşturduğu anlaşılmıştır. Clabin® gibi keratolitik ajanlar bu kaleyi hedef alır; virüsün bulunduğu hiperkeratotik ortamı ortadan kaldırarak enfekte dokuyu katman katman çözer.
6.2. Laktik asidin ve resorsinolün sinerjisi üzerine çalışmalar
Yeni dermatofarmakolojik araştırmalar, laktik asidin stratum korneumu hidrasyonla gevşettiğini, resorsinolün ise keratin proteinlerini daha geçirgen hâle getirdiğini göstermektedir. Bu sinerji, salisilik asidin daha derin tabakalara nüfuz etmesini sağlar.
6.3. Kolodyonun modern rolü
Film oluşturan nitroselüloz sistemleri hâlen güncel araştırmalarda rol oynamaktadır; yapışkan, yarı geçirgen, uzun süreli ilaç salınımı sağlayan bu yapı, Clabin®’in kalıcı başarısının en önemli teknolojik bileşenlerinden biri olmayı sürdürmektedir.
Berotralstat, herediter anjiyoödemin tekrarlayan ataklarının önlenmesi için enzim inhibitörleri grubundan bir aktif maddedir.
Etkileri, bradikinin salınımında rol oynayan serin proteaz plazma kallikreinin inhibisyonuna dayanmaktadır.
Kapsüller günde bir kez yemekle birlikte alınır.
En sık görülen olası yan etkiler karın ağrısı, ishal ve baş ağrısıdır.
Berotralstat, ilaç taşıyıcılarının ve CYP450 izoenzimlerinin bir substratıdır.
Ürünler
Berotralstat kapsül formunda (Orladeyo®) 2020 yılında ABD’de, 2021 yılında AB’de onaylanmıştır.
Yapı ve özellikler
Berotralstat tıbbi üründe, düşük pH’da suda çözünebilen beyaz bir toz olan berotralstat dihidroklorür olarak bulunur.
Etkileri
Berotralstat, serin proteaz plazma kallikreinin bir inhibitörüdür. Bu enzim, damar geçirgenliğini artıran, şişmeye neden olan ve anjiyoödem gelişiminde önemli ölçüde rol oynayan güçlü bir vazodilatör olan bradikinin salınımında rol oynar. Yarılanma ömrü 93 saattir.
Endikasyonlar
Tekrarlayan herediter anjiyoödem ataklarının rutin olarak önlenmesi için.
Ürün bilgilerine göre dozajlanır. Kapsüller günde bir kez yemekle birlikte alınır.
Kontrendikasyonlar
Aşırı Duyarlılık
Tüm önlemler için ürün bilgilerine bakın.
Etkileşimler
Berotralstat, P-glikoprotein ve BCRP ilaç taşıyıcılarının ve CYP2D6 ve CYP3A4’ün bir substratıdır. Aynı zamanda bu CYP izozimlerinin bir inhibitörüdür.
Olumsuz etkiler
En yaygın olası yan etkiler karın ağrısı, ishal ve baş ağrısıdır.
Nabilone, antiemetik özelliklere sahip sentetik bir kannabinoiddir ve öncelikle kanser hastalarında kemoterapinin neden olduğu bulantı ve kusmanın tedavisinde ikinci basamak ajan olarak kullanılır. Etki mekanizması öncelikle merkezi sinir sisteminde dağılmış olan kanabinoid CB1 reseptörleri ile etkileşimine atfedilir. Nabilone yapısal olarak tetrahidrokanabinolün (THC) aktif formu olan dronabinolile ilişkilidir ve esrarla psikoaktif özellikleri paylaşır, bu da onu öforik bir sarhoş edici olarak potansiyel kötüye kullanıma maruz bırakır.
Tarihsel Gelişim ve Ticari Kullanılabilirlik
Nabilone ilk olarak 1970’lerde kannabinoidlerin tıbbi özelliklerine, özellikle de palyatif bakıma olan ilginin arttığı bir dönemde geliştirilmiştir. Ticari kullanılabilirliği**, *Cesamet®* ve Canemes® gibi ticari isimler altında satıldığı ABD, İngiltere ve Almanya dahil olmak üzere birçok ülkeyi kapsamaktadır. Öncelikle kapsül formunda dağıtılır ve psikoaktif potansiyeli göz önüne alındığında bir narkotik ilaç olarak sınıflandırılır. Belirli terapötik kullanımlar için onaylanmış olsa da, daha yaygın olarak tanınan tedavilere kıyasla tıbbi endikasyonlar için sınırlı onay göstererek birçok ülkede ruhsatsız kalmaktadır.
Kimyasal Yapısı ve Özellikleri
Nabilonun** kimyasal formülü C24H36O3 ve molekül ağırlığı 372,5 g/moldür. THC’nin sentetik bir türevidir ve beyaz, kristal toz olarak ortaya çıkar. Farmakolojik özellikleri, THC’ninkiler gibi, hem antiemetik etkilerinden hem de psikoaktif potansiyelinden sorumlu olan CB1 reseptör agonizmini içerir. Nabilonun yaklaşık iki saatlik nispeten kısa bir yarım ömrü vardır, ancak metabolitleri vücutta uzun süre kalabilir ve bu da uzun süreli terapötik veya psikoaktif etkilere katkıda bulunabilir.
Farmakolojik Etkileri ve Etki Mekanizması
Nabilonun antiemetik etkileri öncelikle beyindeki CB1 reseptörleri ile etkileşiminin bir sonucudur. Nörotransmitter salınımını modüle ederek nabilon, özellikle standart antiemetiklere yanıt vermeyen kemoterapi gören hastalarda bulantı ve kusmayı etkili bir şekilde azaltır. Nabilone’un psikoaktif özellikleri, kenevirden elde edilen doğal kannabinoid olan dronabinol (THC) ile benzerdir ve öfori hissi uyandırabilir. Bu etkiler, bazı durumlar için terapötik olarak faydalı olsa da, kötüye kullanım ve bağımlılık ile ilgili endişeleri artırmaktadır.
Endikasyonlar
Nabilone, 5-HT3 reseptör antagonistleri (örn. ondansetron) gibi birinci basamak tedavilerin etkisiz kaldığı durumlarda kemoterapi ile ilişkili bulantı ve kusma tedavisinde öncelikle ikinci basamak ajan olarak endikedir. Onaylanmış endikasyonunun ötesinde, nabilon bazen diğer durumlar için etiket dışı olarak kullanılır:
HIV** hastalarında iştahsızlık ve zayıflama sendromu.
Özellikle palyatif bakım ortamlarında Kronik ağrı yönetimi.
Bununla birlikte, bu endikasyonlar için kullanımı yaygın olarak onaylanmamış veya standartlaştırılmamıştır ve etiket dışı uygulamaları tipik olarak kannabinoid bazlı tedavilere ilişkin daha yumuşak düzenlemelere sahip ülkelerle sınırlıdır.
Kötüye Kullanım Potansiyeli
Psikoaktif özellikleri** göz önüne alındığında, nabilon, esrar veya THC bazlı ürünler gibi öforik bir sarhoş edici olarak kötüye kullanım riskine sahiptir. Öfori**, *rahatlama* ve diğer zihin değiştirici etkiler üretme kabiliyeti, onu kötüye kullanıma açık hale getirir. Bu nedenle, nabilon birçok ülkede kontrollü madde olarak sınıflandırılır ve sporda kullanımı dopingle mücadele düzenlemeleri kapsamında yasaklanmıştır.
Yan Etkiler
Nabilon kullanımıyla ilişkili yan etkiler diğer kannabinoidlerinkine benzerdir ve şunları içerir:
Uyuşukluk**
Baş dönmesi
Ağız kuruluğu
Euphoria
Ataxia (kas koordinasyonu kaybı)
Baş ağrısı
Konsantrasyon güçlüğü
Bu yan etkiler özellikle yüksek dozlarda veya nabilon uzun süre kullanıldığında yaygındır. Psikoaktif** doğası nedeniyle, hastalar günlük işleyişi bozabilecek değişmiş zihinsel durumlar da yaşayabilirler.
Kontrendikasyonlar ve Önlemler
Nabilone, ilaca veya bileşenlerinden herhangi birine karşı bilinen bir aşırı duyarlılığı olan hastalarda kontrendikedir. Psikoaktif etkileri bu durumları daha da kötüleştirebileceğinden, madde bağımlılığı, zihinsel hastalık veya psikotik bozukluk öyküsü olan hastalarda dikkatli kullanılmalıdır. Ek olarak, sedatif özellikleri nedeniyle, hastaların araç kullanma veya ağır makine kullanma gibi dikkat gerektiren faaliyetlerden kaçınmaları önerilir.
Etkileşimler
Nabilon, alkol, benzodiazepinler ve opioidler gibi diğer merkezi sinir sistemi depresanları ile etkileşime girerek potansiyel olarak sedatif etkilerin ve solunum depresyonunun artmasına neden olabilir. Uygulama sırasında dikkatli olunmalıdır.
Etkileşimler
Nabilon, alkol, benzodiazepinler ve opioidler gibi diğer merkezi sinir sistemi depresanları ile etkileşime girerek potansiyel olarak sedatif etkilerin ve solunum depresyonunun artmasına neden olabilir. Bu ilaçlarla birlikte nabilon uygulanırken dikkatli olunmalıdır ve doz ayarlamaları gerekli olabilir.
Dozaj ve Uygulama
Nabilone tipik olarak ürün bilgilerine göre dozajlanarak kapsül formunda uygulanır. Kemoterapiye bağlı bulantı ve kusmada genellikle günde iki kez verilir. Özellikle psikoaktif etkilerine daha duyarlı olabilecek yaşlı veya güçten düşmüş hastalarda yan etki potansiyelini azaltmak için başlangıç dozu genellikle düşüktür.
Keşif
Sentetik bir kannabinoid olan nabilonun geliştirilmesi ve klinik uygulaması, esrarın tıbbi potansiyelinin giderek daha fazla tanınması ve psikoaktif maddelerle ilgili süregelen endişelerle şekillenen büyüleyici bir hikayedir. 1970’lerdeki kökenlerinden bugün palyatif bakımda kullanımına kadar, nabilonun tarihi tıbbi keşif, düzenleme ve kamu algısının kesişimini yansıtmaktadır.
Sentetik Kannabinoidlerin Doğuşu (1970’ler)
1970’ler**, esrarın kemoterapi gören kanser hastalarında *bulantı* ve kusma semptomlarını hafifletebileceğini gösteren anekdot niteliğindeki kanıtların artmasıyla, kannabinoidlerin tedavi potansiyeline yönelik ciddi araştırmaların başlangıcına işaret ediyordu. Bununla birlikte, esrarın yasadışı bir uyuşturucu olarak yasal statüsü tıbbi araştırmalar için zorluklar oluşturuyordu. Bu durum bilim insanlarını, esrarın aktif bileşeni olan tetrahidrokanabinolün (THC) faydalı etkilerini taklit edebilecek sentetik alternatifler aramaya yöneltti.
Nabilone bu dönemde böyle bir sentetik alternatif olarak geliştirildi ve ilk araştırmalar, beyindeki CB1 reseptörleri ile etkileşime girme potansiyelini gösterdi, tıpkı THC gibi, ancak kontrollü, farmasötik bir formda. Nabilonun geliştirilmesi, hastaların çoğu ülkede ağır bir şekilde düzenlenen esrarı doğrudan tüketmeden kannabinoidlerin faydalarını deneyimlemeleri için bir seçenek sağladığından çığır açıcıydı.
Nabilone’un Tanıtımı: Düzenleyici Zorluklar ve Onay (1980’ler)
1980’lerin başlarında**, nabilonun klinik faydaları, özellikle kanser hastaları için zayıflatıcı bir yan etki olan *kemoterapiye bağlı bulantı ve kusmanın* (CINV) tedavisinde daha net hale gelmişti. Bununla birlikte, THC’ye benzer psikoaktif özellikleri ruhsatlandırma onayı alma açısından zorluklar yarattı.
Bu döneme ait bir anekdot, kanser hastalarının nabilon aldıktan sonra sadece bulantılarının azaldığını değil, aynı zamanda öfori ve iyi olma duyguları yaşadıklarını bildirdikleri bir klinik çalışmayı içermektedir. Bu birçok hasta için memnuniyet verici bir rahatlama olsa da, düzenleyiciler arasında kötüye kullanım potansiyeli konusunda endişelere yol açmıştır. ABD Gıda ve İlaç İdaresi (FDA) başlangıçta nabilonu, esrarın etkilerini yansıtan psikoaktif etkileri nedeniyle onaylamakta tereddüt etti. Ancak, diğer ilaçların başarısız olduğu durumlarda bulantıyı kontrol etmedeki etkinliğinin daha ileri çalışmalarla kanıtlanmasının ardından, nabilon nihayetinde 1980’lerin ortalarında bir başka sentetik kannabinoid olan dronabinol ile birlikte kemoterapi kaynaklı bulantı için ikinci basamak tedavi olarak onaylandı.
Etiket Dışı Kullanımlarda ve Kamu Algısında Yükseliş (1990’lar)
1990’larda** nabilonun uygulamaları kemoterapi kaynaklı bulantının ötesine geçmeye başladı. Doktorlar, geleneksel tedavilerin sınırlı başarı sağladığı kronik ağrı ve HIV ile ilişkili anoreksiya gibi diğer durumlar için etiket dışı olarak reçete etmeye başladı. Ağır, kronik hastalıkları olan hastalar nabilonun hem ağrı kesici hem de iştah uyarıcı olduğunu ve daha iyi bir yaşam kalitesi sağladığını keşfetti.
Bu döneme ait ilginç bir anekdot, nabilon aldıktan sonra belirgin bir iştah ve ruh hali iyileşmesi yaşadıkları bildirilen bir grup HIV hastasını içermektedir. Bir hastanın bir röportajda esprili bir şekilde belirttiği gibi, “Günlerce yemek yemezken birdenbire buzdolabındaki her şeyi aşerdim.”
Bununla birlikte, nabilonun artan popülaritesi, keyif verici bir ilaç olarak kötüye kullanımına ilişkin endişeler nedeniyle yasa uygulayıcıları ve düzenleyici kurumlar tarafından artan incelemeyi de beraberinde getirdi. Tıbbi faydaları için değil, esrara benzer öforik etkilerini deneyimlemek için nabilon reçetesi arayan kişilerin raporları ortaya çıktı. Bu durum, birçok ülkede nabilonu kontrollü madde olarak sınıflandıran daha sıkı kontrol ve düzenlemelere yol açtı.
Kannabinoid Bilimindeki Gelişmeler ve Modern Dönem (2000’ler-Günümüz)
2000’li yılların başında endokannabinoid sisteme ve bu sistemin çeşitli fizyolojik süreçleri düzenlemedeki rolüne yönelik ilgi patlamasıyla birlikte kannabinoid araştırmalarında yeni bir dönem başlamıştır. Nabilone, araştırmacıların CB1 reseptörlerini ve bunların ağrı, ruh hali ve iştah düzenlemesi üzerindeki daha geniş etkilerini daha iyi anlamaları için önemli bir araç haline geldi.
Bu dönemde, nabilonun kötüye kullanım potansiyeli önemli bir endişe kaynağı olmaya devam etmiştir. Oldukça kötü şöhretli bir vakada, Birleşik Krallık’ta bir adam, karaborsada esrar yerine satmayı planladığı nabilon kapsüllerini stoklamaktan mahkum edildi. Bu dava, kannabinoidlerin meşru tıbbi kullanımları ile kötüye kullanım riskini dengelemenin süregelen zorluğunu vurgulamıştır.
Bu zorluklara rağmen, nabilone kemoterapiye bağlı bulantı için ikinci basamak tedavi olarak reçete edilmeye devam etti ve doktorlar ondansetron gibi birinci basamak antiemetiklere yanıt vermeyen hastalar için değerini kabul etti. 2010’larda**, *Kanada* ve Almanya dahil olmak üzere birçok ülke, palyatif bakım ortamlarında nabilon kullanımını genişleterek kronik ağrı ve zayıflama sendromlarından muzdarip hastalara rahatlama sağladı.
İleriye Bakmak: Nabilone’un Modern Tıptaki Rolü
Tıbbi araştırmalar kannabinoidlerin potansiyelini keşfetmeye devam ederken, nabilone önemli bir farmasötik araç olmaya devam etmektedir. Son çalışmalar, geleneksel tedavilerin genellikle yetersiz kaldığı fibromiyalji ve nöropatik ağrı gibi durumların tedavisindeki etkinliğini incelemiştir. Psikoaktif özellikleri nedeniyle kontrollü statüsüne rağmen nabilone, sınırlı tedavi seçenekleri olan hastalara umut sunmaktadır.
Nabilonun hikayesi, kanabinoid bazlı tıbbın erken dönemdeki şüphecilik ve düzenleyici zorluklardan tedavi potansiyelinin giderek artan kabulüne kadar geçirdiği geniş evrimi yansıtmaktadır. Tıp camiası kannabinoidlerin karmaşıklığını çözmeye devam ederken, nabilon kenevir türevi tedavilerin faydalarının ihtiyacı olan hastalara yasal ve güvenli bir şekilde ulaşmasını sağlayan yenilikçi çabaların bir kanıtı olarak durmaktadır.
İleri OKuma
Beal, J. E., Olson, R., Laubenstein, L., Morales, J. O., Bellman, P., Yangco, B., … & Shepard, K. V. (1995). Dronabinol as a treatment for anorexia associated with weight loss in patients with AIDS.Journal of Pain and Symptom Management, 10(2), pp. 89-97. DOI: 10.1016/0885-3924(94)00117-4.
Tramer, M. R., Carroll, D., Campbell, F. A., Reynolds, D. J., Moore, R. A., & McQuay, H. J. (2001). Cannabinoids for control of chemotherapy-induced nausea and vomiting: quantitative systematic review.BMJ, 323(7303), pp. 16-21. DOI: 10.1136/bmj.323.7303.16.
Pertwee, R. G. (2006). Cannabinoid pharmacology: the first 66 years.British Journal of Pharmacology, 147(Suppl 1), pp. S163-S171. DOI: 10.1038/sj.bjp.0706406.
Wade, D. T., Makela, P. M., House, H., Bateman, C., & Robson, P. (2006). Long-term use of a cannabis-based medicine in the treatment of spasticity and other symptoms in multiple sclerosis.Multiple Sclerosis Journal, 12(5), pp. 639-645. DOI: 10.1177/1352458506070658.
Abrams, D. I., Jay, C. A., Shade, S. B., Vizoso, H., Reda, H., Press, S., … & Petersen, K. L. (2007). Cannabis in painful HIV-associated sensory neuropathy: a randomized placebo-controlled trial.Neurology, 68(7), pp. 515-521. DOI: 10.1212/01.wnl.0000253187.66183.9c.
Parker, L. A., Rock, E. M., & Limebeer, C. L. (2011). Regulation of nausea and vomiting by cannabinoids.British Journal of Pharmacology, 163(7), pp. 1411-1422. DOI: 10.1111/j.1476-5381.2010.01176.x.
Smith, L. A., Azariah, F., Lavender, V. T., Stoner, N. S., & Bettiol, S. (2015). Cannabinoids for nausea and vomiting in adults with cancer receiving chemotherapy.Cochrane Database of Systematic Reviews, (11), CD009464. DOI: 10.1002/14651858.CD009464.pub2.
Dronabinol, kenevir bitkisi Cannabis sativa L.’nin başlıca psikoaktif bileşeni olan (-)-trans-Δ⁹-tetrahidrokannabinolün (Δ⁹-THC) farmasötik adıdır (C₂₁H₃₀O₂; Mr ≈ 314.5 g/mol). Oda sıcaklığında açık sarı, reçinemsi ve yapışkan bir yağdır; soğukta sertleşir. Yüksek lipofilitesi nedeniyle suda çözünmez; farmasötik çözeltileri tipik olarak susam yağı veya orta zincirli trigliseritlerde (MCT) hazırlanır; yumuşak jelatin kapsüller susam yağı taşıyıcıyla formüle edilir. Susam yağı oksidasyona duyarlı olduğundan ışık/oksijen koruması ve uygun antioksidan/stabilite önlemleri önemlidir. Alman NRF’de magistral formül olarak Özlü Dronabinol Damla %2,5 (NRF 22.8) ve Dronabinol Kapsül 2,5/5/10 mg (NRF 22.7) yer alır; ayrıca inhalasyon amaçlı etanollü 10 mg/mL çözelti (NRF 22.16) tanımlanmıştır.
Etki mekanizması (farmakodinamik)
Δ⁹-THC, endokannabinoid sistemin G-protein eşleşimli reseptörleri olan CB1 (ağırlıkla santral sinir sistemi) ve CB2 (ağırlıkla immün sistem) reseptörlerinde kısmi agonisttir. CB1/CB2, Gi/o üzerinden adenilat siklazı baskılar; voltaj bağımlı Ca²⁺ kanallarını inhibe eder, K⁺ kanallarını aktive eder ve presinaptik nörotransmitter salınımını azaltır. Bu presinaptik inhibisyon, analjezi, antiemetik etki, iştah artışı, kas gevşemesi, anksiyolitik/sedatif etkiler ve psikotropik değişiklikler gibi hem terapötik hem advers sonuçların önemli bir kısmını açıklar.
Farmakokinetik (ADME)
Emilim ve biyoyararlanım
Ağızdan alımda emilim yavaştır ve değişkendir; ilk geçiş metabolizması nedeniyle sistemik biyoyararlanım tipik olarak %10–20 düzeyindedir. Psikoaktif etkiler 30–90 dakika içinde başlar, 2–3 saatte zirveye çıkar ve doza bağlı olarak 4–12 saat sürebilir.
Dağılım
THC yüksek lipofilikliği nedeniyle hızla iyi perfüze dokulara (beyin, kalp, karaciğer) dağılır; ardından yağ dokusunda birikir ve yavaş salınır. Kronik kullanımda yağ dokusundaki birikim, etkilerin ve metabolitlerin saptanabilirliğinin uzamasına katkıda bulunur.
Metabolizma
Karaciğerde başlıca CYP2C9 ve CYP3A4 ile 11-hidroksi-THC (11-OH-THC; aktif) ve 11-karboksi-THC (11-COOH-THC; inaktif) metabolitlerine dönüştürülür. CYP2C9 fonksiyonu azalmış genotiplere sahip bireylerde maruziyet artabilir; dozla ilişkili advers etkiler açısından dikkat gerekir.
Atılım
Metabolitlerin yaklaşık %80–90’ı safra-dışkı yoluyla, %10 kadarı idrarla atılır; terminal yarı ömür çok bölmeli dağılım nedeniyle uzundur. Tek doz sonrası metabolitlerin saptanabilirliği günler ile sınırlıdır; kronik/heavy kullanımda ise haftalarca sürebilir (yağ dokusu depolanması ve yavaş yeniden salınım).
Onaylı endikasyonlar ve kanıt durumu
AİDS ile ilişkili kilo kaybında iştah kaybı (anoreksi): Dronabinol kapsül (Marinol®) ve oral solüsyon (Syndros®) yetişkinlerde endikedir.
Kemoterapiye bağlı bulantı-kusma (KIBK): Konvansiyonel antiemetiklere yanıt vermeyen yetişkinlerde ikinci basamak seçenektir. 2017 Ulusal Bilimler Akademileri raporu, oral kannabinoidlerin KIBK’da etkili antiemetikler olduğuna dair yüksek düzeyde kanıt bildirmiştir.
Kronik/nöropatik ağrı ve spastisite: Çeşitli ülkelerde pratikte kullanılır; etki büyüklüğü orta ve heterojendir, preparat/formülasyona duyarlıdır; ülkelere göre onay durumu değişir.
Regülasyon notu (İsviçre örneği): 1 Ağustos 2022’den itibaren tıbbi amaçlı kenevir yasağı kaldırılmış, olağan narkotik rejimine geçirilmiş; reçete için FOPH istisnai izin zorunluluğu kaldırılmıştır. Benzeri düzenlemeler yargı alanına göre farklılık gösterir.
Oromukozal sprey (nabiksimols, Sativex®): Δ⁹-THC/CBD kombinasyonu; ülkeden ülkeye onay kapsamı değişir (dronabinol değildir). Kanıtlar, oromukozal yolda daha hızlı başlangıç ve daha düşük pik değişkenliği göstermektedir.
Dozlama: etiket temelli şemalar (yetişkin)
AİDS ile ilişkili anoreksi
Kapsül (Marinol®): Başlangıç 2.5 mg günde iki kez, öğle yemeği ve akşam yemeğinden 1 saat önce; tolere edilemiyorsa 2.5 mg yalnızca akşam. Gerektikçe kademeli artır; tipik yanıt 2.5 mg BID; maksimum 10 mg BID.
Oral solüsyon (Syndros®): Başlangıç 2.1 mg günde iki kez (yemeklerden 1 saat önce); tolere edilemiyorsa 2.1 mg akşam; maksimum 8.4 mg BID.
KİBK (konvansiyonel antiemetiklere yanıtsız)
Kapsül (Marinol®): 5 mg/m², kemoterapiden 1–3 saat önce, ardından 2–4 saatte bir, günde 4–6 doz; maksimum 15 mg/m²/doz. İlk doz aç karnına; sonraki dozlar yemekle ilişkili olmaksızın.
Oral solüsyon (Syndros®): 4.2 mg/m², kemoterapiden 1–3 saat önce, sonra 2–4 saatte bir, günde 4–6 doz; maksimum 12.6 mg/m²/doz. İlk doz aç karnına ≥30 dk önce; sonraki dozlar yemekle ilişkili olmaksızın.
Doz titrasyonu “low and slow” ilkesiyle yapılmalı; santral sinir sistemi (SSS) advers etkileri izlendiğinde ara veya küçük adımlarla ayarlama önerilir. Yaşlılar için daha düşük başlangıç dozu tercih edilir.
Klinik etkiler ve başlangıç-süre dinamikleri
Ağızdan alımda etkiler genellikle 30–90 dk içinde başlar; psikotrop etkiler 2–3 saatte tepe yapar ve 4–12 saat sürebilir. İştah uyarımı ve bazı somatik etkiler daha uzun sürebilir; bireyler arası değişkenlik büyüktür.
Kontrendikasyonlar ve uyarılar
Aşırı duyarlılık: Dronabinol veya formül bileşenlerine (ör. susam yağı; oral solüsyon için alkol/PG) karşı. Syndros: Disülfiram veya metronidazol ile eş zamanlı/son 14 gün içinde kullanım kontrendikedir (disülfiram-benzeri reaksiyon riski).
Nöropsikiyatrik etkiler: Anksiyete, disfori/euforia, bilişsel bozulma, psikotik belirtiler tetiklenebilir; psikiyatrik öyküsü olanlarda kaçınılmalı ya da sıkı izlenmelidir. Araç/makine kullanımı etkilenir.
Hemodinamik etkiler: Taşikardi, ortostatik hipotansiyon/senkop; kardiyak hastalığı olanlarda dikkat ve vital izlem önerilir. (
Nöbet riski: Dronabinol ile nöbet/nöbet-benzeri durumlar bildirilmiştir; risk-yarar değerlendirmesi gerektirir.
Gebelik/laktasyon: Fetal zarar potansiyeli nedeniyle kaçınılmalıdır; emzirme önerilmez.
Bağımlılık ve yoksunluk: Kontrollü maddedir (ABD’de kapsül CIII, oral solüsyon CII). Kesilince irritabilite, uykusuzluk, iştah değişiklikleri vb. yoksunluk semptomları olabilir.
İlaç etkileşimleri
Farmakokinetik: Güçlü CYP2C9/3A4 inhibitörleri (örn. flukonazol, ketokonazol, makrolidler, ritonavir, greyfurt suyu) maruziyeti artırabilir; indükleyiciler (örn. rifampisin) azaltabilir. Genetik CYP2C9 zayıf metabolizörlerde maruziyet artışı görülebilir. Syndros etanollü olduğundan disülfiram/metronidazol ile birlikte verilmez.
Farmakodinamik: Alkol, benzodiazepinler, barbitüratlar, opioidler, sedatif antidepresanlar ve diğer SSS baskılayıcılarla aditif sedasyon ve reaksiyon yavaşlaması; antikolinerjiklerle taşikardi artışı olasıdır. Dar terapötik indeksli ve yüksek protein bağlanımlı ilaçlarla (ör. varfarin, siklosporin, amfoterisin B) yer değiştirme ve toksisite riski nedeniyle yakın izlem önerilir.
Advers etkiler (dozla ilişkili ve sıklıkla erken dönemde)
Çok sık/sık: Baş dönmesi, somnolans, öfori/dysfori, “high” hissi, paranoid reaksiyon, düşünme bozukluğu, iştahsızlık veya artışı, abdominal ağrı, bulantı/kusma, ağız kuruluğu, çarpıntı/taşikardi, yüzde kızarma, vazodilatasyon. Daha seyrek/ciddi: Hipotansiyon/senkop, hipertansiyon, psikotik atak/halüsinasyon, konfüzyon, dezoryantasyon, nöbet, düşme, aşırı duyarlılık reaksiyonları.
Özel popülasyonlar
Geriatrik: Nöropsikiyatrik ve postural hipotansif etkilere daha duyarlı; düşük başlangıç dozu ve yavaş titrasyon önerilir.
Hepatik yetmezlik: Metabolizma karaciğerde olduğundan dikkat ve doz ayarı gerekebilir (etiket: dikkat uyarıları).
Tedavi başlangıcı ve doz artırımlarında mental durum, kan basıncı/nabız, postüral belirtiler ve düşme riski izlenmelidir.
AİDS anoreksisinde vücut ağırlığı ve iştah günlüğü; KİBK’de emesis sayısı ve kusma-karşıtı kurtarma ihtiyacı takip edilmelidir.
Alkol ve SSS baskılayıcıları ile eşzamanlı kullanımdan kaçınma; araç/makine kullanmama uyarısı zorunludur.
Toksikoloji ve adli/atletik hususlar
THC-COOH idrar eşiği (WADA) 150 ng/mL’dir; bu değerin üstündeki müsabaka içi numuneler pozitif kabul edilir (Karar Limiti ~180 ng/mL). Sporcularda dopingle mücadele kuralları ve lig/organizasyon politikaları ayrıca değişebilir.
Galenik ve stabilite (eczacılık uygulamaları)
Yağ çözeltileri (MCT veya susam yağı) amber şişelerde, oksidasyona karşı koruyucu koşullarda saklanmalıdır. Susam yağı taşıyıcılı kapsüller oksidasyona duyarlı olabilir; raf ömrü etiket/NRF rehberleriyle belirlenir. Etanollü inhalasyon çözeltileri (NRF 22.16) yalnızca uygun cihaz/şartlarda ve belirtildiği endikasyonlarda kullanılmalıdır.
Uygulamada yer alan ürünler ve erişim
ABD’de Marinol® (kapsül) ve Syndros® (oral solüsyon) mevcuttur (etiketleri ayrıntılı doz talimatı ve uyarıları içerir). Birçok Avrupa ülkesinde eczaneler magistral dronabinol preparatları hazırlayabilir (NRF tarifeleri). Yargı alanlarına göre onay, geri ödeme ve reçeteleme koşulları değişir; İsviçre’de 1 Ağustos 2022 düzenlemeleriyle hekim reçetelemesi idari izin gerektirmeden mümkün hale gelmiştir.
Keşif
Dronabinol’un (−)-trans-Δ⁹-tetrahidrokannabinol olarak kimlik kazanması, 1964’te yayımlanan kısa ama yüksek yoğunluklu bir makaleyle—Gaoni ve Mechoulam’ın haşhaşişten tek bir psikoaktif ilkeyi izole edip, yapısını aydınlatıp ve kısmen sentezleyerek doğruladıkları çalışmayla—bilim tarihine sabitlendi. Bu tarih bir “ilk bulma”dan fazlasıdır: kenevir kimyasının yüzyıl başından beri süren dağınık ipuçlarını bir eksen etrafında toplayan bir dönüm noktasıdır. O ana dek kenevir, oksijenle temas ettikçe koyulaşan, güneşte kıvam değiştiren, çözeltideyken bile kimyagerin elinden sıyrılan bir reçineydi; kimyasal özne değil, karmaşık bir matristi. Dronabinol’ün keşfi, bu matrisi bir molekül ciddiyetine indirgeyerek farmakolojide ölçülebilir doz–yanıt, eczacılıkta ölçeklenebilir formülasyon ve tıpta denetlenebilir endikasyon kapılarını açtı.
Bu kırılmanın arka planında, 20. yüzyılın ilk yarısında atılan ama birbirini tamamlamayan adımlar durur. 1899’da cannabinol (CBN) reçineden ayrıştırıldığında, kenevirin “aktif ruhu” olduğuna inanılan bu bileşiğin aslında oksidatif bir son ürün olduğu yeterince anlaşılamamıştı; yıllar sonra Δ⁹-THC’nin havada, ışık ve ısıyla CBN’ye yönelen kaderi, önceki kuşakların niçin hep “başka bir yere” ulaştığını açıklayacaktı. 1940’ta Adams ve arkadaşlarının cannabidiol’ü (CBD) izole etmesi, “psikoaktif olmayan ikiz”in varlığını kanıtlayarak tabloyu yeni bir eksene oturttu. Fakat asıl düğüm, can alıcı halkaların nasıl bağlandığı, çift bağın nerede durduğu ve stereokimyanın ne olduğuyuydu: Kısacası hangi molekül sorusu, 1963’te CBD’nin yapısı çözüldüğünde nihai hamlesini bekleyen bir satranç konumuna dönüşmüştü.
1964 hamlesinin gücü, üçlü kanıt zincirinde yatar. Birincisi, izolasyon: Polis kaynaklı haşhaşiş numunelerinden (o yıllarda başka türlü erişmek neredeyse imkânsızdı) tek bir etkin prensip—hafif sarı, reçinemsi, son derece lipofilik bir yağ—farmasötik titizlikle ayrıldı. İkincisi, yapısal aydınlatma: O günün imkânlarıyla (UV, IR, kütle spektrumu, dönme açısı; erken dönem NMR) halkalı iskelet, yan zincirin uzunluğu ve çift bağın konumu gösterildi; literatürde o sıralar “Δ¹” diye anılan bağ, günümüz terpene numaralandırmasına çevrildiğinde “Δ⁹” olarak yerini aldı. Üçüncüsü, kısmi sentez: CBD’den yola çıkan kimyasal dönüşümlerle elde edilen ürünün, doğal kaynaklı bileşenden ayrıştırılamaması yapının doğruluğunu mühürledi. Bu “izolasyon + yapı + sentez” üçlemesi, farmasötik kimyanın altın standardıdır; tek başına hiçbirinin ikna ediciliği, üçünün birlikteki ağırlığına ulaşmaz.
Keşfin niçin zor olduğuna dair kimyasal gerekçeler, klasik bir laboratuvar güncesi gibi okunur. Δ⁹-THC aşırı lipofiliktir; suya neredeyse yabancı, yağda ise fazlaca “evinde”dir. Böyle bir molekülü reçine karışımından çekip almak, çok basamaklı kromatografi ve oksidasyona karşı disiplin ister. Ayrıca molekül ısı ve ışığa duyarlıdır; doğru yapılmayan her işlemin ödülü, sizi yine cannabinol’e yaklaştıran bir oksidasyon olur. Bir başka incelik, stereokimya ve nomenklatürdür: Eski kayıtlardaki “Δ¹-THC”, bugün (−)-trans-Δ⁹-THC’dir; bilimsel hafıza, numaralandırma sistemleri arasındaki çeviri işini ciddiye almazsa, tarihi yanlış okuyabilir.
Molekül tanımlanır tanımlanmaz iki şey mümkün oldu: Bir, mekanizma dili doğdu. Daha sonraki yıllarda beynin CB₁ reseptörüne bağlanan kısmi agonist bir ligand olarak tasvir edilecek bu yapı, presinaptik nörotransmitter salınımını baskılayan bir biyolojinin kimyasal simgesi hâline geldi; ağrı, mide bulantısı, iştah ve kas tonusu gibi farklı klinik fenomenler ortak bir endokannabinoid eksende açıklanabilir oldu. İki, formülasyon aklı yerleşti. Lipofilik bir molekülü mağistra yağ damlalarına, yumuşak jelatin kapsüllere, ileride etanollü oral çözeltilere dönüştürmek; başlangıç–zirve–süre dinamiklerini öngörmek; ilk geçiş metabolizmasıyla barışık doz stratejileri geliştirmek artık mümkün hâle geldi. Dronabinol böylece 1980’lerden itibaren kemoterapiye bağlı bulantı-kusma ve AİDS ile ilişkili anoreksi gibi alanlarda kontrollü doz–yanıt eğrileriyle sahneye çıktı; klinik farmakolojide “yavaş başla, yavaş artır” prensibiyle psikotrop ve hemodinamik etkiler yönetilebilir bir çerçeveye oturdu.
Bu biyolojik dilin klinik karşılığı, 1980’lerin ortasından itibaren ete kemiğe büründü. Ağızdan alınan bir molekül olarak dronabinolün en zorlayıcı özelliği—yüksek ilk geçiş metabolizması ve değişken emilim—klinik farmakolojinin ritmini belirledi: Etkilerin 30–90 dakika içinde başlaması, 2–3 saatte tepe yapması, psikotrop titreşimlerin 4–12 saat sürmesi; bunun yanında lipofilik depo alanları nedeniyle etkilerin ve metabolitlerin izinin günlerce sürmesi. Bu kinetik tablo, kemoterapiye bağlı bulantı-kusmanın atak paternine uygun bir dozlama stratejisi (tedavi öncesi yükleme ve 2–4 saatte bir tekrarlama) ile AIDS ilişkili anorekside daha tonik bir iştah uyarımı hedefleyen iki farklı klinik “müzik”e dönüştü. Kapsül formu, susam yağı taşıyıcıda nispeten sabit bir emilim profili sunarken; yıllar sonra gelen oral solüsyon, besin–alkol–yardımcı çözücü etkileşimlerini daha hassas yönetmeyi gerektiren ama doz ayarlamasını incelten bir seçenek sundu. Magistra damlalar (özellikle MCT/susam yağlı preparatlar) ise eczacılık pratiğinde bireyselleştirilmiş titrasyona imkân vererek “low and slow” yaklaşımını güçlendirdi.
Reseptör düzeyi keşiflerin klinik güvenlilikle örüldüğü dönemde, sistemin sınırları da belirginleşti. Dronabinolün CB₁’de kısmi agonist oluşu, onu sentetik “tam agonist” dalgasından (piyasada “Spice/K2” gibi adlarla yayılan, toksisite profili sert bileşikler) ayırdı; yine de psikotrop etkiler—anksiyete ve disforiden öfori ve paranoid epizotlara uzanan ölçek—ve hemodinamik dalgalanmalar (taşikardi, postural hipotansiyon) doz ve bireyler arası değişkenliğe duyarlı kaldı. Bu yüzden klinik kılavuzlar, özellikle yaşlı hastalarda düşük başlangıç dozu, yavaş titrasyon, düşme riski yönetimi ve eş zamanlı SSS baskılayıcılarından kaçınma gibi “ritüelleri” zorunlu kıldı. Farmakogenomik mercekle bakıldığında CYP2C9 varyantlarının maruziyeti artırabildiği anlaşıldı; bu da “kimya defterindeki küçük notların” klinikte gerçek karşılıkları olduğunu gösterdi.
Toplumsal ve düzenleyici sahnede ise hikâye zikzaklı bir seyre sahipti. Bir yanda tıbbi endikasyonlar için kapılar aralanırken, diğer yanda kötüye kullanım ve güvenlik başlıkları farklı ülkelerde farklı hızlarla yönetildi. Spor dünyası örneğinde, eşik değerlerin yıllar içinde revize edilmesi, molekülün rekabetçi alanlardaki konumunu daha gerçekçi bir çerçeveye oturttu. Avrupa’da 2010’lardan itibaren oromukozal THC/CBD kombinasyon spreyinin (dronabinol olmayan ama aynı eksene ait bir hazırlık) çok ülkeli onayları, kanıta dayalı ama ölçülü bir genişleme anlamına geldi; eczanelerin magistra yetkinliği olan ülkelerde (örneğin Alman NRF tarifeleriyle) yağ damlaları ve kapsüller, bireyselleştirilmiş tedavinin araçlarına dönüştü. İsviçre’nin 2022’de tıbbi kullanım için idari izni kaldırıp hekim reçetesini yeterli sayan reformu, “kenevirin tıpta normalleşmesi” başlığının sembollerinden biri oldu.
Formülasyon bilimi bu süreçte sürekli çalıştı: Susam yağının oksidasyona duyarlılığını azaltacak saklama koşulları ve ambalajlar, MCT taşıyıcılarda çözünürlük–stabilite dengeleri, oral çözeltilerde yardımcı çözücülerin hasta eğitimiyle birlikte yönetilmesi; hepsi uzun bir pratik aklın ürünüdür. Klinik düzlemde ise iki sabit nokta hiç değişmedi: Doz–yanıt ilişkisini tekil bir “mükemmel sayı”ya indirgememek ve hastayı—psikiyatrik öykü, kardiyovasküler durum, eşzamanlı ilaçlar—ile birlikte okumak. AİDS ilişkili anoreksi ve kemoterapiye bağlı bulantı-kusmada gelen düzenleyici onayların arkasında, işte bu “kimyadan kliniğe” akışın sayısız küçük düzeltmesi vardır.
İleri Okuma
Wood TB, Spivey WTN, Easterfield TH. (1899). Cannabinol. Part I. Journal of the Chemical Society, Transactions, 75: 20–36.
Adams R, Hunt M, Clark JH. (1940). Isolation of cannabidiol, a constituent of Cannabis sativa L. Journal of the American Chemical Society, 62(1): 196–200.
Mechoulam R, Shvo Y. (1963). Hashish—Part I: The structure of cannabidiol. Tetrahedron, 19(12): 2073–2078.
Gaoni Y, Mechoulam R. (1964). Isolation, structure, and partial synthesis of an active constituent of hashish. Journal of the American Chemical Society, 86(8): 1646–1647.
AbbVie Inc. (güncellemeli ilk onay 1985). MARINOL® (dronabinol) Capsules — Prescribing Information. U.S. FDA Label.
Devane WA, Dysarz FA, Johnson MR, Melvin LS, Howlett AC. (1988). Determination and characterization of a cannabinoid receptor in rat brain. Molecular Pharmacology, 34(5): 605–613.
Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. (1990). Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature, 346(6284): 561–564.
Devane WA, Hanuš L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, et al. (1992). Isolation and structure of a brain constituent that binds to the cannabinoid receptor (anandamide). Science, 258(5090): 1946–1949.
Munro S, Thomas KL, Abu-Shaar M. (1993). Molecular characterization of a peripheral receptor for cannabinoids (CB₂). Nature, 365(6441): 61–65.
Hoffman AF, Lupica CR. (2000). Mechanisms of Cannabinoid Inhibition of GABA_A Synaptic Transmission in the Hippocampus. Journal of Neuroscience, 20(7): 2470–2479.
Grotenhermen F. (2003). Pharmacokinetics and Pharmacodynamics of Cannabinoids. Clinical Pharmacokinetics, 42(4): 327–360.
National Academies of Sciences, Engineering, and Medicine. (2017). The Health Effects of Cannabis and Cannabinoids: The Current State of Evidence and Recommendations for Research. The National Academies Press. (NCBI)
AbbVie Inc. (2017). MARINOL® (dronabinol) Capsules — Full Prescribing Information. FDA Label, Rev. 08/2017. (FDA Access Data)
Lucas CJ, Galettis P, Schneider J. (2018). The Pharmacokinetics and the Pharmacodynamics of Cannabinoids. British Journal of Clinical Pharmacology, 84(11): 2477–2482. (PMC)
WADA. (2022). 2023 Prohibited List – Explanatory Notes (Cannabis threshold 150 ng/mL; DL 180 ng/mL). World Anti-Doping Agency. (World Anti Doping Agency)
Swissmedic. (2022). Amendments to Narcotics Ordinances on Cannabis for Medical Purposes (effective 1 Aug 2022). Swissmedic Notice. (Swissmedic)
Benuvia Operations, LLC. (2024). SYNDROS® (dronabinol) Oral Solution — Full Prescribing Information. FDA Label, Rev. 05/2024. (FDA Access Data)
Kendi kendine deneyde, genellikle yeni geliştirilen veya keşfedilen bir aktif farmasötik bileşen veya yeni bir tıbbi ürün, genellikle bir bilim insanının kendisi tarafından denenir. Amaç, farmakolojik özellikler hakkında hızlı bir şekilde daha fazla bilgi edinmektir.
Geçmişte düzenli olarak kendi kendine denemeler gerçekleştirilmiştir. En iyi bilinen örneklerden biri Albert Hofmann’ın 19 Nisan 1943’te LSD ile yaptığı ve “LSD – mein Sorgenkind” adlı kitabında anlattığı kendi deneyidir. Hofmann çok yüksek dozda 0.25 mg LSD almış ve bu da yoğun halüsinasyon deneyimine yol açmıştır. Bu deney, Hofmann’ın kendini iyi hissetmediği için sarhoşken işten eve bisikletle dönmesi nedeniyle de bilinir. Bu deney başarılı oldu çünkü LSD’nin psikotropik etkisi çok belirgindir ve en küçük dozlarda bile ortaya çıkar.
Günümüzde kendi kendine deneyler artık ilaç geliştirme bağlamında ya da sadece zararsız maddelerle veya farmakolojik etkileri bilinen maddelerle yapılmamaktadır. Bunun nedeni, örneğin zehirlenme ve aşırı doz gibi bariz risklerdir. Bilinmeyen aktif maddeler de kanserojen ve teratojen olabilir ve prosedür etik açıdan sorgulanabilir.
Ayrıca, plasebo kontrollü bir test olmadığı ve genellikle sadece bir denek dahil olduğu için anlamlılık sınırlıdır.
Demir oksitler ilaç, gıda ve kozmetikte kırmızı, sarı, kahverengi ve siyah boyalar olarak kullanılır. Eczacılıkta genellikle tabletler ve kapsüller için kullanılırlar.
Ürünler
Demir oksitler özel mağazalarda saf maddeler olarak mevcuttur. Birçok tıbbi üründe, özellikle tablet ve kapsüllerde yardımcı madde olarak bulunurlar.
Yapısı ve özellikleri
Demir oksitler, inorganik bileşikler (pas) olarak bulunan demir oksitleridir. Renklendirici olarak kullanılan çeşitli temsilciler vardır. Suda çözünmeyen kırmızı, kahverengi, sarı ve siyah tozlar halinde bulunurlar. Bu maddeler demir hidroksitler de içerebilir.
Demir oksit kırmızısı (Kırmızı demir oksit): Fe2O3
Demir oksit sarısı (Sarı demir oksit): Fe2O3 – H2O (monohidrat)
Demir oksit siyahı (Siyah demir oksit): Fe3O4
Uygulama alanları
Gıda, ilaç ve kozmetik için renklendirici olarak. Demir oksitler ayrıca birbirleriyle ve diğer boyalarla (örneğin titanyum dioksit) karıştırılır. Ayrıca ilaçlar için mürekkep olarak ve UV ışınlarını absorbe etmek için kullanılırlar.
İstenmeyen etkiler
Demir oksitlerin öngörülen miktarlarda zararsız olduğu kabul edilir.
Bromelain, ananas bitkisinin sap ve meyvelerinden elde edilen ve çeşitli proteolitik enzimler içeren bir preparattır. Bromelain, diğerlerinin yanı sıra anti-enflamatuar, anti-ödematöz, fibrinolitik, antitümör ve antitrombotik özelliklere sahiptir.
Şişlik, enflamasyon ve ağrı için, ayrıca steroid olmayan anti-enflamatuar ilaçlara ve glukokortikoidlere bitkisel bir alternatif olarak uygulanır.
Tabletler açken, yemekten yarım saat ila bir saat önce alınır. Dozlama aralığı preparata bağlıdır.
En yaygın olası yan etkiler alerjik reaksiyonlar ve gastrointestinal rahatsızlıkları içerir.
Bromelain diğer ilaçların emilimini artırabilir ve antikoagülanlar ve antiplatelet ajanlar ile kombine edilmemelidir.
Ürünler
Bromelain kaplanmış tabletler (Traumanase®) şeklinde ticari olarak mevcuttur ve ananas tozu içeren diyet takviyeleri mevcuttur. Wobenzym® 2022 yılında yeni tescil edilmiştir. Aynı yıl, yanık kabuklarının giderilmesine yönelik bir jel de onaylanmıştır (NexoBrid®).
Kimyasal
Yapı ve özellikler
Bromelain, ananas bitkisinin (Ananas comosus, Bromeliaceae) saplarından (meyve sapları) ve meyvelerinden elde edilen ve çeşitli proteolitik enzimler içeren bir preparattır. Proteolitik olmayan bileşenler de bu etkide rol oynar. Diğer bitkilerin aksine, enzimler olgun meyvelerde hala uygun bir konsantrasyonda bulunmaktadır.
Etkileri
Bromelain anti-enflamatuar, anti-ödematöz, fibrinolitik, immünomodülatör, antitümör, antimikrobiyal, antiplatelet ve antitrombotik özelliklere sahiptir.
Endikasyonlar
Belirgin ödemli yumuşak doku enflamasyonunda yardımcı olarak.
Yaralanmalar sonucu şişme, iltihaplanma veya ağrı.
İdrar ve genital sistem iltihabı
Yüzeysel flebit
Osteoartrit
Yumuşak doku romatizması
Topikal jel:
Derin termal yaralanmaları olan yetişkinlerde yanık kabuğunun çıkarılması için (NexoBrid®). Diğer endikasyonlar literatürde tanımlanmıştır. Bunlar arasında özellikle kanser yer almaktadır. Bromelain ayrıca NSAID’lere ve glukokortikoidlere bitkisel bir alternatif olarak da kullanılmaktadır.
Gıda teknolojisinde bromelain, papain gibi eti yumuşatmak için kullanılır.
Ürün bilgilerine göre dozajlnaır. Tabletler açken, yemekten yarım saat ila bir saat önce alınır. Dozaj aralığı ilaca göre değişir.
Kontrendikasyonlar
Aşırı Duyarlılık
Kan pıhtılaşma bozuklukları
Antikoagülanların veya antiplatelet ilaçların birlikte uygulanması.
Karaciğer ve böbrek hastalıkları
Ameliyattan önce
Çocuklar
Hamilelik ve emzirme (tavsiye edilmez)
Tüm önlemler ilaç bilgi broşüründe bulunabilir.
Etkileşimler
Bromelain diğer tıbbi ürünlerin emilimini artırabilir ve özellikle antibiyotiklerin (örn. tetrasiklinler, sülfonamidler, amoksisilin) plazma seviyelerini yükseltebilir. Antikoagülanlar ve trombosit agregasyon inhibitörleri ile artmış bir kanama eğilimi göz ardı edilemez.
Olumsuz etkiler
En sık görülen olası yan etkiler alerjik reaksiyonlar ve gastrointestinal şikayetlerdir.
Araştırmalar, “Dulaglutid”in muhtemelen “iki GLP-1 molekülü” anlamına geldiğini, “dula”nın “duo”dan ve “glutid”in de glukagon benzeri peptid-1’den geldiğini ve dimerik yapısını yansıttığını gösteriyor.
Kanıtlar, “Trulicity”nin, açık bir kamusal kökeni olmayan, muhtemelen pazarlanabilirlik için seçilmiş Eli Lilly’nin bir marka adı olduğu yönünde.
Dulaglutid’in Etimolojisi
“Dulaglutid”, tip 2 diyabet tedavisinde kullanılan bir ilacın genel adıdır. İsmin kimyasal yapısından, özellikle insan immünoglobulin G4’ün bir Fc parçasına bağlı iki GLP-1 (glukagon benzeri peptid-1) analoğuna sahip dimerik bir molekül olmasından türetilmiş olması muhtemeldir. “Dula” öneki, iki anlamına gelen “duo”dan geliyor olabilir ve dimerik yapıyı gösterirken, “glutid”in kan şekeri kontrolünde rol oynayan bir hormon olan GLP-1 ile ilişkili olması muhtemeldir.
Trulicity’nin Etimolojisi
“Trulicity”, Eli Lilly tarafından pazarlanan Dulaglutide’nin marka adıdır. “Trulicity”nin kökeni kamuya açık olarak belirtilmemiştir, ancak pazarlama çekiciliği için “true” ve “elicity” gibi unsurları bir araya getirerek oluşturulmuş bir isim gibi görünmektedir, doğrudan bilimsel bir anlamı yoktur.
Dulaglutide’in Etimolojisi
“Dulaglutide” jenerik adı, ilacın moleküler yapısını ve işlevini yansıtan bilimsel olarak türetilmiş gibi görünmektedir. Diğer GLP-1 agonistleri (örneğin, exenatide, liraglutide, semaglutide, albiglutide) gibi benzer ilaçlarla ilgili araştırmalar, farmasötik isimlerin genellikle kimyasal bileşimlerinin veya etki mekanizmalarının unsurlarını içerdiğini göstermektedir.
İsmin Ayrımı: “Dulaglutid”, “dula” ve “glutid” olarak ayrılabilir. “Glutid” eki GLP-1 agonistlerinde yaygındır, muhtemelen “glukagon benzeri peptit”ten türetilmiştir, “glu” glikozla ilgilidir ve “tide” bir peptidi belirtir. Örneğin, albiglutid de “glutid” kullanır ve “albi” öneki, kaynaştığı albüminle ilgilidir.
Dimerik Yapı: Dulaglutid, küçük bir peptit zinciri aracılığıyla insan IgG4’ün bir Fc parçasına bağlı iki özdeş GLP-1 analog dizisinden oluşan bir dimerik molekül olarak tanımlanır. Bu, “dula”nın iki anlamına gelen “duo”dan geldiğini ve ikili GLP-1 yapısını vurguladığını düşündürmektedir. Bu yorum, Dulaglutide – DrugBank Online gibi kaynaklarda ayrıntılı olarak açıklandığı gibi, ilacın biyokimyasal yapısıyla uyumludur.
İşlevsel Alaka: Kan şekeri kontrolünü iyileştirmek için GLP-1’i taklit etmedeki rolü göz önüne alındığında, isim muhtemelen hem yapısını (dimerik) hem de işlevini (GLP-1 agonisti) vurgulamaktadır ve bu da “Dulaglutide”i tanımlayıcı bir jenerik isim haline getirmektedir.
Trulicity’nin Etimolojisi
Marka adı olarak “Trulicity”, genellikle pazarlanabilirlik, akılda kalıcılık ve yasal bulunabilirlik için seçilen farklı bir adlandırma kuralını takip eder. Jenerik isimlerin aksine, marka isimlerinin mutlaka bilimsel bir temeli yoktur ve genellikle ilaç pazarında öne çıkmak için oluşturulurlar.
Kamu Kaynağının Olmaması: Ticari marka başvuruları ve şirket duyuruları da dahil olmak üzere kapsamlı aramalar, “Trulicity”nin nasıl seçildiğine dair açık ayrıntıları ortaya koymamıştır. Ticari marka, Eli Lilly tarafından 23 Kasım 2010’da, TRULICITY Ticari Marka Başvurusu‘de görüldüğü gibi, diyabet tedavisi için farmasötik preparatlar için dosyalandı, ancak etimolojik bir açıklama sağlanmadı.
Olası Yorum: Spekülatif olarak, “Trulicity” “true” (güvenilirlik veya etkililik anlamına gelir) ve “elicity” (muhtemelen insülin salınımını uyarmadaki ilaç eylemiyle ilgili olan “elicit” kelimesinden türemiştir) kelimelerini birleştirebilir. Ancak, marka adları genellikle keyfi olduğu ve markalama amaçları için tasarlandığı için bu çıkarımsaldır ve doğrulanmamıştır.
Pazarlama Bağlamı: Eli Lilly’nin diyabet bakımına odaklanması göz önüne alındığında, “Trulicity” güven ve basitliği çağrıştırmak için seçilmiş olabilir ve hasta uyumunu amaçlayan haftada bir enjeksiyon için uygundur. Bu, isimlerin tüketici algısı ve yasal bulunabilirlik açısından test edildiği ilaç markalama stratejileriyle tutarlıdır.
Diğer GLP-1 Agonistleriyle Karşılaştırmalı Analiz
Bağlamlandırmak için, diğer GLP-1 agonistleri isimlendirme kalıplarına ilişkin içgörü sağlar:
Eksenatid (Byetta): Gila canavarının exendin-4’ünden türetilmiştir, “exen” muhtemelen “ekzokrin” anlamına gelir.
Liraglutid (Victoza): “Lira” yine Gila canavarı araştırmasından “kertenkele” ile ilişkili olabilir.
Semaglutid (Ozempic): “Sema” reseptör aktivasyonunu yansıtan “sinyal” anlamına gelebilir.
Albiglutid (Tanzeum): Albüminden “Albi”, GLP-1 ile kaynaşmıştır, “glutid” sınıf boyunca tutarlıdır.
Bu kalıp, “glutid”in GLP-1 agonistleri için bir sınıf tanımlayıcısı olduğu ve Dulaglutid’deki “dula”nın dimerik doğasını benzersiz bir şekilde yansıtarak onu diğerlerinden ayırdığı hipotezini destekler.
Sınıflandırma ve Yapı Dulaglutid, glukagon benzeri peptid-1 (GLP-1) reseptör agonist sınıfına ait uzun etkili bir antidiyabetik ajandır. Doğal insan GLP-1 ile yaklaşık %90 amino asit dizisi homolojisini paylaşan iki özdeş zincir içeren genetik olarak tasarlanmış bir füzyon proteinidir. Amino asitlerin ikamesi ve bir immünoglobulin G4 Fc fragmanı ile füzyon dahil olmak üzere yapısal modifikasyonlar, endojen GLP-1’i doğal olarak inaktive eden dipeptidil peptidaz-4 (DPP-4) enzimi tarafından bozunmaya karşı direnci artırır.
Farmakokinetik Bu modifikasyonlar, yaklaşık 5 günlük uzatılmış bir yarılanma ömrü sağlayarak haftada bir kez subkutan uygulamaya olanak tanır. Bu uzun süreli etkinlik, dozlama sıklığını azaltarak daha kısa etkili GLP-1 agonistlerine kıyasla hasta bağlılığını artırır.
Etki Mekanizması Dulaglutid, GLP-1 reseptörlerine bağlanır ve bunları aktive ederek glukoza bağlı etkiler gösterir:
Glukagon Baskılanması: Pankreatik α-hücrelerinden yemek sonrası glukagon salgılanmasını inhibe eder.
Doygunluk Artışı: Mide boşalmasını yavaşlatır ve tokluğu teşvik etmek için merkezi olarak hareket eder.
Daha da önemlisi, glukoza bağlı etkisi, insülin veya sülfonilürelerde yaygın bir endişe olan hipoglisemi riskini en aza indirir.
Klinik Kullanım 2014 yılında ABD FDA ve 2015 yılında EMA tarafından onaylanan dulaglutide, genellikle diyet, egzersiz ve diğer glikoz düşürücü ajanlara ek tedavi olarak tip 2 diabetes mellitus yönetiminde endikedir. Trulicity®* (Eli Lilly and Company) markası altında pazarlanmaktadır.
Güvenlik ve Tolerabilite Yaygın yan etkiler gastrointestinaldir (örn. bulantı [~%12-21], diyare [~%9-17], kusma ve karın ağrısı), tipik olarak geçici ve doza bağımlıdır. Nadir fakat ciddi riskler arasında pankreatit ve tiroid C-hücresi tümörü endişeleri bulunmaktadır (kişisel/ailesel medüller tiroid karsinomu veya multipl endokrin neoplazi sendromu tip 2 öyküsünde kontrendikedir). Güvenlik profili, endojen incretin hormonlarının etkisini taklit eden diğer incretin mimetikleri ile uyumludur.
Uygulama şekli Subkutan enjeksiyon için önceden doldurulmuş, tek dozluk bir kalem olarak mevcuttur (haftalık 0,75 mg veya 1,5 mg), diyabet yönetimi protokollerinde kolaylık ve esneklik sunar.
Keşif
Araştırmalar Dulaglutide’in keşfinin 2005 yılı civarında bir patent başvurusuyla başladığını göstermektedir.
İlk klinik denemelerin 2006’da, Faz III denemelerin ise 2010’da başlaması muhtemel görünüyor.
Kanıtlar 2014 yılında FDA onayına, 2020 yılında ise genişletilmiş kullanım onayına işaret etmektedir.
Keşif ve Erken Gelişim
Tip 2 diyabet için bir ilaç olan Dulaglutide, muhtemelen 2005 yılı civarında önemli bir patentin [US 7,452,966 patent pdf] dosyalanmasıyla işaretlenen keşif aşamasına sahipti (https://patentimages.storage.googleapis.com/pdfs/US7452966.pdf). Bu dönem, klinik deneyler için zemin hazırlayan ilk sentez ve preklinik çalışmaları içermektedir.
Klinik Çalışmalar ve Onay
İlk klinik çalışmalar, muhtemelen Faz I, 2006 yılında başlamış ve sağlıklı gönüllülerde güvenlik ve farmakokinetik konularına odaklanmıştır. Daha büyük gruplarda etkinliği test eden Faz III denemeleri 2010 yılı civarında başlamış ve olumlu sonuçlar 2013 yılında açıklanmıştır. FDA, Dulaglutide’i Eylül 2014’te tip 2 diyabetin yönetimi için onayladı ve 2020’de, başlangıçtaki diyabet tedavisinin ötesinde beklenmedik bir genişleme ile daha yüksek dozlar ve kardiyovasküler riskleri azaltmak için onaylandı.
Beklenmedik Detay
İlginç bir ayrıntı da Dulaglutide’in majör advers kardiyovasküler olayları azaltmak için onaylanan ilk tip 2 diyabet ilacı olması ve kan şekeri kontrolünün ötesinde hasta sağlığı üzerindeki daha geniş etkisini vurgulamasıdır.
Dulaglutide Hakkında Arka Plan
Dulaglutide, rekombinant DNA teknolojisi kullanılarak tasarlanmış, tip 2 diyabet yönetiminde diyet ve egzersize ek olarak onaylanmış, uzun etkili bir GLP-1 reseptör agonistidir. İlk olarak Eylül 2014’te FDA tarafından onaylanmış ve daha sonra Şubat 2020’de tip 2 diyabet ve kardiyovasküler risk faktörleri olan hastalarda majör advers kardiyovasküler olayları (MACE) azaltmayı içerecek şekilde genişletilmiştir. Geliştirilmesi, biyolojik ilaçlar için tipik bir yörüngeyi yansıtan sentezden klinik deneylere ve ruhsat onayına kadar birçok aşamayı içeriyordu.
Keşif ve Geliştirme Zaman Çizelgesi
Dulaglutide’in keşfi ve geliştirilmesine ilişkin zaman çizelgesi, patent başvuruları, klinik deney kayıtları ve şirket duyurularından çıkarılan birkaç önemli kilometre taşına ayrılabilir:
2005: Keşif aşaması muhtemelen bu tarihlerde başlamıştır. 29 Eylül 2005 tarihinde Dulaglutide’in yapısı, işlevi, üretimi ve tip 2 diyabet tedavisinde kullanımını detaylandıran US 7,452,966 numaralı patent başvurusu yapılmıştır. Bu patent, 18 Kasım 2008 tarihinde verilmiş olup, ilk araştırma ve sentezin 2000’li yılların ortalarında gerçekleştiğini ve diğer GLP-1 agonistlerinin gelişimi ile aynı hizada olduğunu göstermektedir.
2006: İlk klinik çalışmalar, özellikle Faz I, NCT00327655 olarak tanımlanan en erken çalışma ile başladı; bu çalışma Dulaglutide’in sağlıklı deneklerde farmakokinetiğini, farmakodinamiğini ve güvenliğini değerlendirmek için Mayıs 2006’da başladı. Bu, insanlarda güvenliğe odaklanan preklinik gelişimden klinik gelişime geçişi işaret etmektedir.
2008-2010: Belirli başlangıç tarihleri daha az belgelenmiş olsa da, Faz II denemeleri muhtemelen bu dönemde gerçekleşmiştir. Bu denemeler, tip 2 diyabetli hastalarda doz bulma ve daha ileri güvenlik değerlendirmelerini içerecek ve daha büyük Faz III çalışmalarına hazırlanacaktı. Daha sonraki yayınlarda görülen kesintisiz Faz 2/3 çalışma tasarımı, potansiyel olarak 2008 civarında başlayan uyarlanabilir deneme tasarımlarının kullanıldığını göstermektedir.
2010-2012: Faz III klinik çalışmalar başlamış, tanımlanan en erken çalışma olan NCT01075282 (AWARD-2) Mart 2010’da başlamıştır. Bu çalışma, metformin ve glimepirid kullanan hastalarda Dulaglutid ile insülin glargini karşılaştırmıştır ve birincil tamamlanma tarihi Ocak 2012’dir. AWARD-1 (NCT01064687, Şubat 2010’da başlıyor) gibi diğer AWARD denemeleri de başlamış ve çeşitli karşılaştırıcılara karşı etkinliği değerlendirmiştir. Bu denemeler, daha geniş popülasyonlarda etkinliğin gösterilmesi açısından çok önemliydi.
2013: Eli Lilly, 15 Nisan 2013’te AWARD-2 ve AWARD-4 adlı iki ek Faz III çalışmasının olumlu sonuçlarını açıkladı ve insülin glargine kıyasla hemoglobin A1c seviyelerini düşürmede başarılı sonuçlar elde edildiğini gösterdi. Bu dönüm noktası, ilacın pazar onayı potansiyelini gösteren ruhsatlandırma başvurusu için kritik öneme sahipti.
2014: FDA, 18 Eylül 2014 tarihinde AWARD programına dayanarak tip 2 diyabetli yetişkinlerde glisemik kontrolü iyileştirmek için Dulaglutide’i onayladı. Bu onay, pankreatit ve potansiyel medüller tiroid karsinomu gibi riskleri yönetmek için bir Risk Değerlendirme ve Azaltma Stratejisi (REMS) içeriyordu. Avrupa Birliği de Kasım 2014’te onay vererek küresel erişimi genişletmiştir. 2020: Şubat 2020’de FDA, REWIND çalışma sonuçlarına dayanarak Dulaglutide’in endikasyonlarını tip 2 diyabetli ve yerleşik kardiyovasküler hastalığı veya birden fazla risk faktörü olan yetişkinlerde MACE riskini azaltmayı içerecek şekilde genişletti. Ayrıca, Eylül 2020’de, AWARD-11 çalışmasına dayanarak, glisemik kontrol ve kilo yönetimi için daha fazla seçenek sunan daha yüksek dozlar (3.0 mg ve 4.5 mg) onaylanmıştır. Diğer GLP-1 Agonistleri ile Karşılaştırmalı Analiz Dulaglutide’in zaman çizelgesini bağlamsallaştırmak için diğer GLP-1 reseptör agonistleri ile bir karşılaştırma yapmak faydalı olacaktır:
Bu tablo, Dulaglutide’in geliştirme zaman çizelgesinin diğer GLP-1 agonistleriyle tutarlı olduğunu, muhtemelen bir Fc fragmanıyla kaynaşmış yeni dimerik yapısı nedeniyle klinik çalışmalara girmeden önce biraz daha uzun bir klinik öncesi aşamaya sahip olduğunu vurgulamaktadır.
Kesin Tarihlerin Belirlenmesindeki Zorluklar Keşif ve erken geliştirme için kesin tarihlerin belirlenmesi, klinik öncesi araştırmaların kamuya açıklanmasının sınırlı olması nedeniyle zorluklarla karşılaşmıştır. 2005’teki patent başvurusu net bir işaret sağlarken, kesin sentez tarihi ve ilk klinik öncesi çalışmalar çıkarımsaldır. clinicaltrials.gov adresindeki klinik araştırma verileri Faz I ve III araştırmaları için kesin başlangıç tarihleri sağlamıştır, ancak Faz II araştırmalarının kesin zaman çizelgeleri daha az belgelenmiştir ve tipik ilaç geliştirme aşamalarına dayalı tahminler gerektirmektedir.
Beklenmedik Detay: Kardiyovasküler Faydalar İlginç bir ayrıntı da Dulaglutide’in 2020’de MACE’yi azaltmak için onay alması ve hem birincil hem de ikincil önleme popülasyonları için böyle bir endikasyona sahip ilk tip 2 diyabet ilacı olmasıdır. REWIND çalışmasına dayanan bu genişleme, diyabet yönetiminde önemli bir ilerleme olan glisemik kontrolün ötesinde daha geniş terapötik potansiyelinin altını çizmektedir.
Tablo: Önemli Klinik Denemeler ve Önemli Noktalar
Önemli Nokta/Olay
Tarih
Açıklama
Patent Başvurusu (US 7,452,966)
Eylül 2005
İlk keşif ve geliştirme aşamasını işaret ediyor.
Birinci Aşama I Denemesi (NCT00327655)
Mayıs 2006
Sağlıklı deneklerde farmakokinetik, farmakodinamik ve güvenliği değerlendirdi.
Faz III Denemeleri Başlıyor (AWARD-2)
Mart 2010
Büyük ölçekli etkinlik ve güvenlik denemelerine başlandı, örn. NCT01075282.
Olumlu Faz III Sonuçları Duyuruldu
Nisan 2013
Eli Lilly, AWARD-2 ve AWARD-4’ten etkinlik gösteren sonuçları duyurdu.
T2DM için FDA Onayı
Eylül 2014
REMS uygulamasıyla tip 2 diyabette glisemik kontrol için onaylandı.
CV Faydaları için Genişletilmiş Onay
Şubat 2020
Kardiyovasküler risk faktörleri olan hastalarda MACE’yi azaltmak için onaylandı.
Daha Yüksek Dozlar Onaylandı
Eylül 2020
AWARD-11 deneme sonuçlarına göre 3,0 mg ve 4,5 mg dozları onaylandı.
Bu tablo, Dulaglutide’in keşiften pazara sunulmasına kadar olan yolculuğunun kronolojik açıdan net bir genel görünümünü sunarak kritik dönüm noktalarını özetlemektedir.
İleri Okuma
Barrington, P., Chien, J.Y., Tibaldi, F., Showalter, H.D., Schneck, K., & Ellis, B. (2011). LY2189265, a long-acting glucagon-like peptide-1 analogue, showed a dose-dependent effect on insulin secretion in healthy subjects.Diabetes, Obesity and Metabolism, 13(5), 434–438.
Umpierrez, G.E., Povedano, S.T., Manghi, F.P., Shurzinske, L., & Pechtner, V. (2014). Efficacy and safety of dulaglutide monotherapy vs metformin in type 2 diabetes in a randomized controlled trial (AWARD-3).Diabetes Care, 37(8), 2168–2176.
Wysham, C., Blevins, T., Arakaki, R., Colon, P., Garcia, P., Atisso, C., & Lakshmanan, M.C. (2014). Efficacy and safety of dulaglutide added on to pioglitazone and metformin versus exenatide in type 2 diabetes in a randomized controlled trial (AWARD-1).Diabetes Care, 37(8), 2159–2167.
Nauck, M., Weinstock, R.S., Umpierrez, G.E., Guerci, B., Skrivanek, Z., Milicevic, Z., & Forst, T. (2014). Efficacy and safety of dulaglutide versus sitagliptin after 52 weeks in type 2 diabetes in a randomized controlled trial (AWARD-5).Diabetes Care, 37(8), 2149–2158.
Dungan, K.M., Povedano, S.T., Forst, T., González, J.G., Atisso, C., Sealls, W., & Fahrbach, J.L. (2014). Once-weekly dulaglutide versus once-daily liraglutide in metformin-treated patients with type 2 diabetes (AWARD-6): a randomised, open-label, phase 3, non-inferiority trial.The Lancet, 384(9951), 1349–1357.
Zhang, L., Zhang, M., Zhang, Y., & Tong, N. (2016). Efficacy and safety of dulaglutide in patients with type 2 diabetes: a meta-analysis and systematic review.Scientific Reports, 6, 18904.
Tuttle, K.R., Lakshmanan, M.C., Rayner, B., Busch, R.S., Zimmermann, A.G., & Long, J. (2018). Dulaglutide versus insulin glargine in patients with type 2 diabetes and moderate-to-severe chronic kidney disease (AWARD-7): a multicentre, open-label, randomised trial.The Lancet Diabetes & Endocrinology, 6(8), 605–617.
Gerstein, H.C., Colhoun, H.M., Dagenais, G.R., Diaz, R., Lakshmanan, M., Pais, P., Probstfield, J., Riddle, M.C., Ryden, L., Xavier, D., & Yale, J.F. (2019). Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): a double-blind, randomised placebo-controlled trial.The Lancet, 394(10193), 121–130.
Gerstein, H.C., Sattar, N., Rosenstock, J., Ramasundarahettige, C., Pratley, R., Lopes, R.D., Lam, C.S.P., Heenan, L., Del Prato, S., & Yale, J.F. (2020). Cardiovascular and renal outcomes with dulaglutide in type 2 diabetes across categories of age and baseline risk: a prespecified analysis of the REWIND trial.The Lancet Diabetes & Endocrinology, 8(7), 505–516.
Solomon, S.D., Uno, H., Lewis, E.F., Eckardt, K.U., Komajda, M., McMurray, J.J.V., Maggioni, A., Martinez, F.A., Packer, M., Pfeffer, M.A., Swedberg, K., & Zannad, F. (2021). Dulaglutide and heart failure outcomes in patients with type 2 diabetes: an analysis from the REWIND trial.Circulation, 143(12), 1173–1183.






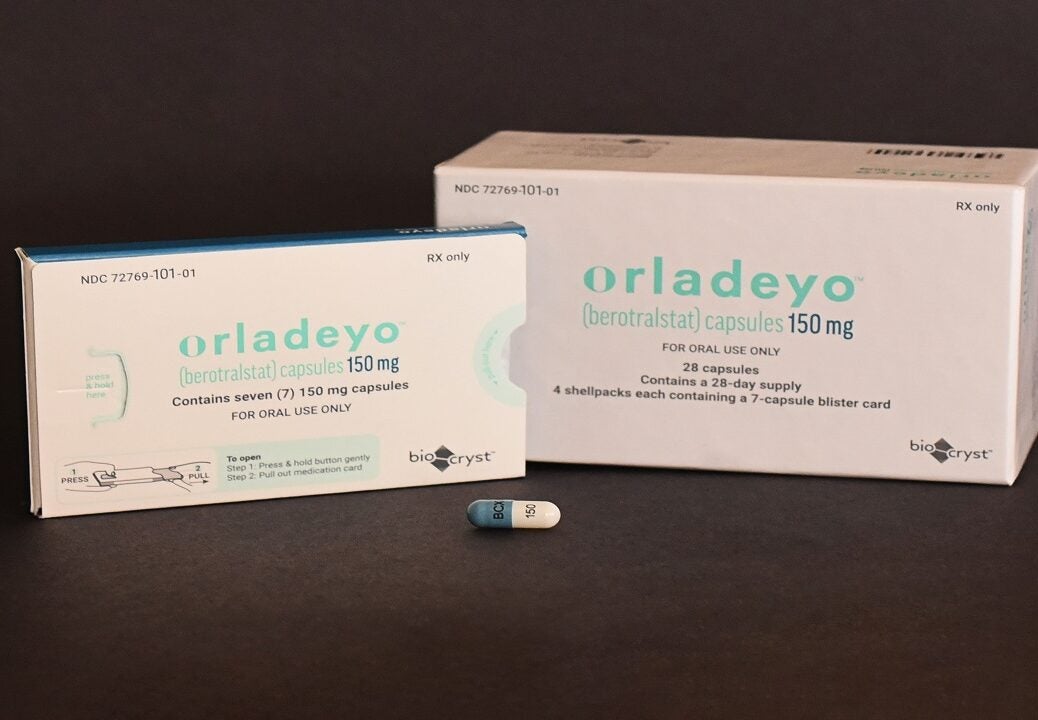










Yorum yazabilmek için oturum açmalısınız.