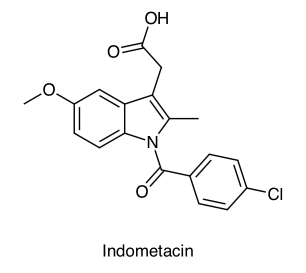
Steroid olmayan iltahap engelleyicilerdendir. İltahap azaltıcı, ateş düşürücü, ağrı azaltıcı etkisi vardır. Etkisi Prostaglandinlerin biosentezini engellemesinden kaynaklanır.
Tıp terimleri sözlüğü

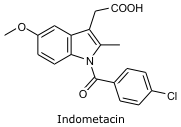
Steroid olmayan iltahap engelleyicilerdendir. İltahap azaltıcı, ateş düşürücü, ağrı azaltıcı etkisi vardır. Etkisi Prostaglandinlerin biosentezini engellemesinden kaynaklanır.

Sinonim: Erythromycin
Aslında Streptomyces erythreus tarafından üretilir.Antibiyotik işlevinden dolayı ilaç olarak kullanılır.

Fusidik asit terimi, bir yandan bileşiğin ilk elde edildiği mantarın cins adına (Fusidium) doğrudan göndermede bulunurken, diğer yandan organik kimyada yaygın olan “-ik asit” adlandırma geleneğini taşır. “Fusid-” kökü bu nedenle etken maddenin filogenetik ya da yapısal sınıflandırmasından çok, keşif-bulunuş bağlamına işaret eden tarihsel bir imzadır. Nitekim fusidik asit, kimyasal olarak “steroid benzeri” görünümüne rağmen, klasik anlamda memeli steroid hormonlarıyla biyosentetik akrabalıktan ziyade, mantarların ürettiği tetrasitklik triterpenoid bir iskelet (fusidan çekirdeği) üzerinde yükselen özgün bir antibiyotiktir. Bu ikili karakter—steroidal mimariyi andıran bir iskelet ile antibiyotik işlevin tek bir molekülde birleşmesi—hem adlandırmayı hem de klinik algıyı biçimlendirmiştir: fusidik asit, antibiyotikler içinde “hedefe kilitlenen” mekanizmasıyla ayırt edilirken, “steroid antibiyotik” etiketi nedeniyle zaman zaman kortikosteroidlerle karıştırılmaya açık bir kavramsal gölge de taşır.
Fusidik asit, 20. yüzyılın ortasında doğal ürün antibiyotiklerinin altın çağında, Fusidium coccineum kültürlerinden izole edilerek literatüre girdi. Bu dönem, penisilinlerin ve sefalosporinlerin klinik sahneyi dönüştürdüğü, streptomisin ve tetrasiklinlerin endikasyon alanlarını genişlettiği; buna karşılık özellikle stafilokokların hızla direnç geliştirdiği bir ekosistemdi. Dolayısıyla yeni bir antistafilokokal ajanın keşfi, yalnızca “yeni bir antibiyotik” değil, aynı zamanda hastane enfeksiyonları ve cilt-yumuşak doku enfeksiyonları yönetiminde stratejik bir araç anlamına geliyordu. Zaman içinde fusidik asit, Avrupa’da çeşitli formlarıyla yaygınlaştı; bazı ülkelerde topikal kullanım, bazılarında sistemik kullanım daha belirgin bir yer edindi. Klinik tarihteki kalıcı rolünün ardında, hedefinin (EF-G) antibiyotikler arasında görece özgün olması, stafilokoklara karşı güçlü aktivitesi ve uygun seçilmiş olgularda iyi bir doku penetrasyonu profili yatmaktadır.
Tarihsel çizgide dikkat çekici bir nokta şudur: fusidik asit, “geniş spektrumlu” olma iddiasından ziyade “hedeflenmiş spektrum” mantığıyla değer kazanmıştır. Antibiyotik tarihinin önemli bir kısmı geniş spektruma doğru bir genişleme hikâyesi iken, fusidik asit daha çok belirli bir patojen grubunu (özellikle Staphylococcus aureus) baskılamada uzmanlaşmış bir ajan olarak konumlanmıştır. Bu durum, modern antimikrobiyal yönetim (stewardship) düşüncesiyle de uyumludur: gereksiz geniş spektrum kullanımını sınırlamak, hedefe yönelik tedaviyi güçlendirmek ve ekolojik yan etkileri azaltmak.
Fusidik asit, mantarların mikrobiyal rekabette kullandığı ikincil metabolitlerden biridir. Toprak ve organik madde ekosistemlerinde mantarlar, bakterilerle aynı nişleri paylaşır; besin, alan ve kaynak için rekabet eder. Bu rekabetin evrimsel sonucu, antibakteriyel moleküllerin—özellikle ribozom ve protein sentezi gibi “yaşamın çekirdeği” süreçleri hedefleyen bileşiklerin—çeşitlenmesidir. Fusidik asidin hedefi olan elongasyon faktörü G (EF-G), bakteriyel translasyonun vazgeçilmez motor proteinlerinden biridir; bu hedefin seçilmesi, mantar açısından yüksek getirili bir stratejidir: bakterinin yaşamsal bir düğüm noktasına müdahale edilir.
Ancak evrimsel yarış tek yönlü değildir. Bakteriler, fusidik aside karşı başlıca iki eksende karşı-savunma geliştirir:
Bu çerçeve, modern klinik pratiğe şu mesajı taşır: fusidik asit, hedefi özgün olduğu için değerli olsa da, tek başına ve özellikle yüksek bakteri yükü içeren, yaygın veya invaziv enfeksiyonlarda direnç seçilimi açısından dikkatle yönetilmelidir. Evrimsel biyolojinin diliyle söylersek, her antibiyotik uygulaması bir seçilim deneyidir; fusidik asit de bu deneyin güçlü bir seçici baskı unsuru olabilir.
Fusidik asit, steroid benzeri tetrasitklik bir iskelet taşıyan, yüksek derecede lipofilik bir moleküldür. Bu lipofilik karakter, membranlarla etkileşim, doku dağılımı ve formülasyon tercihlerinde belirleyicidir. Serbest asit formu suda düşük çözünürlük gösterirken, sodyum tuzu (sodyum fusidat) suda belirgin daha iyi çözünür; bu nedenle bazı sistemik ve özellikle parenteral formülasyonlarda tuz formu tercih edilir. Klinik pratikte en yaygın formlar topikal preparatlardır (krem, pomad/merhem, jel, emdirilmiş gazlı bez vb.); bazı ülkelerde oral formlar (tablet/süspansiyon) ve daha sınırlı bağlamda parenteral seçenekler de yer alır.
Formülasyon çeşitliliği yalnızca pazarlama çeşitliliği değildir; farmakolojik bir gerekçesi vardır: fusidik asidin etkin olduğu enfeksiyon alanları sıklıkla cilt ve yumuşak dokudur, ayrıca kemik-eklem enfeksiyonları gibi “antibiyotiğin hedef dokuya ulaşmasının zor olduğu” tablolar da endikasyon yelpazesinde tartışılır. Topikal formülasyonlar lokal yüksek konsantrasyon avantajı sağlar; sistemik formlar ise derin doku enfeksiyonlarında gerekir, ancak sistemik etkileşim ve yan etki yükünü de beraberinde getirir.
Fusidik asidin farmakodinamik kimliği, bakteriyel translasyonun elongasyon aşamasındaki kritik bir adımı hedeflemesine dayanır. EF-G, ribozom üzerinde GTP hidroliziyle enerji sağlayan bir translokazdır: tRNA’ların ve mRNA’nın ribozom içindeki konum değişimini (translokasyon) hızlandırır ve aynı zamanda ribozom geri dönüşümü süreçlerinde rol alır. Fusidik asit, EF-G’nin ribozomla etkileşiminin belirli bir konformasyonel evresinde bağlanır ve EF-G’nin ribozomdan ayrılmasını engeller. Sonuç, “çalışan makinenin kilitlenmesi” gibidir: ribozom EF-G ile birlikte takılı kalır, bir sonraki elongasyon çevrimi başlayamaz, protein sentezi işlevsel olarak durur.
Bu mekanizma iki nedenle klinik açıdan değerlidir:
Mikrobiyolojik düzeyde fusidik asidin etkisi çoğu durumda bakteriyostatik olarak tanımlanır; buna rağmen yüksek konsantrasyonlarda veya belirli koşullarda bakterisidal etki gözlenebilir. Klinik karar açısından önemli olan, etkinliğin yalnızca “öldürme” hızına değil, hedef patojene karşı yeterli maruziyetin sağlanmasına ve direnç seçiliminin yönetilmesine bağlı olduğudur.
Fusidik asit, ağırlıklı olarak gram-pozitif bakterilere karşı etkindir; klinik pratiğin merkezinde stafilokoklar bulunur. Metisiline duyarlı S. aureus (MSSA) üzerinde güçlü aktivite klasik bir özelliktir; bazı metisiline dirençli S. aureus (MRSA) izolatlarında da aktivite korunabilir, ancak bu, yerel direnç paternlerine ve mekanizmalara bağlıdır. Koagülaz-negatif stafilokoklar ve bazı streptokok türleri de duyarlılık gösterebilir. Gram-negatif bakterilerde dış membran bariyeri ve hedef erişilebilirliği gibi nedenlerle etkinlik sınırlıdır; bu nedenle fusidik asit “geniş spektrum” amaçla seçilmez.
Klinik olarak öne çıkan kullanım alanları şunlardır:
Bu noktada kritik ilke şudur: fusidik asidin özellikle sistemik kullanımında monoterapi, direnç seçilimini kolaylaştırabileceği için dikkatle değerlendirilmelidir. Bazı klinik rehber yaklaşımları, yüksek riskli tablolar veya yüksek bakteri yükü durumlarında kombinasyon stratejilerini öne çıkarır; karar, enfeksiyonun lokalizasyonu, ağırlığı, patojenin duyarlılık profili ve hastanın eşlik eden durumlarıyla birlikte verilmelidir.
Fusidik aside direnç gelişimi, klinik açıdan iki ana nedenle önem taşır: birincisi, özellikle topikal antibiyotiklerin yaygın ve bazen kontrolsüz kullanımı yerel seçilim baskısını artırabilir; ikincisi, EF-G hedefli direnç mutasyonları bakterinin fizyolojisini etkileyebildiği için “direnç-uygunluk” dengesi zaman içinde değişebilir.
Direnç mekanizmaları klinik pratikte şu sonuçlara yol açabilir:
Fusidik asidin lipofilik doğası, farmakokinetik davranışını belirgin biçimde şekillendirir:
Farmakokinetik, yalnızca “vücutta ne olur” sorusu değildir; doğrudan güvenlik ve etkileşim yönetiminin temelidir. Fusidik asit, klinik pratikte özellikle statinlerle birlikte kullanımda ciddi kas toksisitesi olasılığı nedeniyle dikkat çekmiştir; bu etkileşim, mekanizması çok etmenli olsa da, sistemik fusidik asit kullanımı planlanan hastalarda statin tedavisinin geçici olarak kesilmesi veya alternatif antibiyotik seçimi gibi stratejileri gündeme getirir.
Fusidik asit, karaciğer metabolizması ve taşıyıcı sistemler üzerinden çok sayıda ilaçla klinik açıdan anlamlı etkileşim potansiyeli taşır. Pratikte en kritik senaryolar şunlardır:
Etkileşim yönetimi, yalnızca “kaçın” komutuna indirgenemez; bazen enfeksiyonun ciddiyeti fusidik asidi gerekli kılar. Bu durumda alternatif ilaç seçimi, doz ayarı, geçici kesme stratejileri ve laboratuvar/klinik izlem, rasyonel bir risk azaltma paketinin parçalarıdır.
Topikal fusidik asit, özellikle yüzeyel stafilokokal cilt enfeksiyonlarında tercih edilir. Uygulama sıklığı (günde 2–3 kez gibi) klinik uygulamada yaygındır; ancak ideal yaklaşım, mümkün olan en kısa süreyle ve sınırlı alana uygulamadır. Geniş alanlara, uzun süreli ve tekrarlayan kürler halinde uygulama, direnç seçilimini kolaylaştırabilir. Oklüzif pansuman altında kullanım, lokal konsantrasyonu artırabilir; buna bağlı irritasyon ve duyarlanma reaksiyonları da artabileceğinden dikkat gerektirir.
Oral veya parenteral kullanım, daha derin enfeksiyonlarda gündeme gelir. Doz, endikasyon, patojen duyarlılığı, hastanın karaciğer fonksiyonu ve eşzamanlı ilaçlarına göre belirlenir. Sistemik kullanımda iki ilke öne çıkar:
Topikal preparatlar genellikle iyi tolere edilir; buna rağmen lokal yanma, batma, kaşıntı, eritem ve kontakt dermatit görülebilir. Bariyer bütünlüğü bozulmuş ciltte emilim artabileceği için hem lokal irritasyon hem de teorik sistemik maruziyet artışı dikkate alınmalıdır.
Sistemik kullanımda gastrointestinal yakınmalar (bulantı, kusma, ishal, abdominal rahatsızlık) sık bildirilir. Karaciğer enzimlerinde yükselme ve kolestatik sarılık gibi hepatobilier yan etkiler klinik açıdan önemlidir; özellikle uzun süreli kullanımda veya eşzamanlı hepatotoksik ilaç kullanımında izlem gerektirir. Nadir fakat klinik olarak ağır olabilen reaksiyonlar arasında aşırı duyarlılık tabloları ve belirgin kas toksisitesi riskinin arttığı etkileşim senaryoları yer alır.
Yan etki yönetimi, hastanın risk profilinin önceden belirlenmesiyle başlar: karaciğer hastalığı, ileri yaş, polifarmasi, statin kullanımı, böbrek fonksiyon bozukluğuna eşlik eden komplikasyonlar ve önceki ilaç reaksiyon öyküsü değerlendirilmelidir.
Fusidik asit, “eski” bir antibiyotik olmasına karşın bilimsel gündemden düşmüş değildir. Güncel araştırma çizgileri üç eksende toplanabilir:

Bu bağlamda fusidik asit, antibiyotik keşfinin yalnızca “yeni molekül bulma” değil, mevcut moleküllerin biyolojiyle daha iyi hizalanması, direnç ekolojisinin yönetilmesi ve hedef odaklı tedavinin güçlendirilmesi olduğunu hatırlatan bir örnektir.
1950’lerin sonunda antibiyotik araştırması, bir yandan penisilinin açtığı çağın ivmesini sürdürürken, öte yandan stafilokokların giderek daha “inatçı” hale geldiği klinik gerçeklikle yüzleşiyordu. Hastanelerde yara enfeksiyonları, cerrahi alan enfeksiyonları ve cilt-yumuşak doku tabloları içinde Staphylococcus aureus giderek daha sık “tedaviye direnç gösteren” bir aktör olarak öne çıkıyor; araştırmacılar ise doğanın kimyasal çeşitliliğinin hâlâ bilinmeyen ceplerinde yeni silahlar bulunabileceği umuduyla tarama programlarını genişletiyordu. Bu dönemde Danimarka’da, Kopenhag çevresindeki endüstriyel araştırma ekosisteminin önemli bir parçası olan LEO Laboratuvarları (sonraki adıyla LEO Pharma) içinde yürütülen programların bir hattı, toprağın sessiz biyokimyasına kulak kesildi: mikroorganizmaların birbirini baskılamak için ürettiği ikincil metabolitler, klinikte işe yarayabilecek “doğal antibiyotik” adaylarıydı.
Hikâyenin başlangıcı, laboratuvarın rutin ama sabır isteyen bir pratiğinde yatar: toprak örneklerinin toplanması, içlerindeki mantar ve bakterilerin ayrıştırılması, saf kültürlerin kurulması ve her bir kültür filtratının (veya ekstraktının) hedef bakterilere karşı inhibisyon gücünün taranması. 1959 yılında LEO araştırmacıları, Fusidium coccineum olarak tanımlanan bir mantar kültüründen elde edilen materyalin özellikle gram-pozitif bakteriler üzerinde belirgin bir baskı oluşturduğunu fark etti. Bu, ilk bakışta “yeni bir inhibitör” sinyaliydi; fakat antibiyotik keşfinde asıl mesele, sinyalin gerçek bir moleküler varlığa dönüştürülmesidir: etkinliği taşıyan bileşiğin izolasyonu, saflaştırılması, yapısal karakterizasyonu ve tekrar tekrar aynı biyolojik etkiyi verdiğinin gösterilmesi.
Bu aşama, çoğu zaman görünmez kahramanlıkların devresidir. Çözücü ekstraksiyonları, kristalizasyon denemeleri, kromatografik ayrımlar… Her adımda aktiviteyi kaybetmemek gerekir; çünkü biyolojik etkinlik, saflaştırma boyunca “iz” olarak takip edilen tek pusuladır. Nihayet, beyaz kristal bir madde elde edilir: daha sonra “fusidik asit” adıyla anılacak olan molekül.
Keşfin akademik dünyaya sağlam bir giriş yapabilmesi için, yalnızca “bulduk” demek yetmez; bileşiğin temel özellikleri, etkinlik spektrumu ve klinik değer potansiyeli ortaya konmalıdır. 1962’de W. O. Godtfredsen liderliğinde, S. Jahnsen, H. Lorck, K. Roholt ve L. Tybring ile birlikte yayımlanan çalışma, fusidik asidi bilim dünyasına net bir kimlikle tanıttı. Buradaki dönüştürücü nokta şuydu: fusidik asit, sıradan bir “yeni antibiyotik” değildi; steroid benzeri tetrasitklik bir iskeleti andıran özgün mimarisi nedeniyle, klasik antibiyotik ailelerinin (beta-laktamlar, aminoglikozidler, tetrasiklinler vb.) dışında, farklı bir kimyasal evrenden geliyordu. Kimyasal “görünüş”, farmakolojik “davranış” için her zaman garanti olmasa da, araştırmacıların zihninde güçlü bir merak uyandırır: Bu kadar farklı bir iskelet, bakteriyi nasıl durduruyordu?
Godtfredsen ekibi ve çağdaşları, ilk klinik umudu da aynı netlikle işaretledi: gram-pozitiflere, özellikle stafilokoklara karşı güçlü aktivite. Bu vurgu tesadüf değildir; o yıllarda stafilokok enfeksiyonları, modern hastane pratiğinin en can yakıcı sorunlarından biridir. Bir keşif, ancak klinik bir boşluğa dokunabiliyorsa “önemli” sayılır; fusidik asit o boşluğa dokunuyordu.
1960’ların ilk yarısı, fusidik asidin bir yandan farmasötik geliştirme sürecine, diğer yandan klinik gözlemlere taşındığı dönemdir. Molekülün lipofilik karakteri ve serbest asit formunun düşük suda çözünürlüğü, daha uygun farmasötik formlara—özellikle sodyum tuzu formlarına—yönelişi teşvik eder. Bu yıllarda klinisyenlerin ve endüstriyel araştırmacıların ortak hedefi, etkinliği korurken uygulanabilirliği artırmaktır: topikal preparatlar, cilt enfeksiyonlarına odaklı bir pratik kolaylık sağlar; sistemik formlar ise daha derin dokulara uzanma ihtimalini doğurur.
Bu dönemde literatüre giren katkılar, çoğu zaman “büyük mekanizma” sorusuna cevap vermekten ziyade, ilacın organizma düzeyindeki etkilerini, tolerabilitesini ve farmakolojik izlerini anlamaya yöneliktir. Böylece fusidik asit, keşif anındaki “parlak aktivite” sinyalinden, klinik reçeteye doğru uzanan bir olgunlaşma çizgisi kazanır.
Fusidik asidi özgün kılan yönlerden biri, bakteriyi durdurma biçiminin klasik ribozom inhibitörlerinden ayrışmasıdır. 1960’ların sonuna gelindiğinde araştırmacılar, fusidik asidin bakteriyel protein sentezini etkilediğini giderek daha güçlü biçimde sezmeye başlar; fakat “nerede” ve “nasıl” soruları, moleküler biyolojinin o günkü araçlarıyla sabırla yanıtlanacaktır.
1969’da yayımlanan çalışmalar, fusidik asidin ribozom–G faktörü (sonradan daha net terminolojiyle uzama faktörü G, EF-G) ekseninde bir “yakalama/kilitleme” etkisi oluşturduğuna dair ikna edici biyokimyasal işaretler üretir. Buradaki kavrayış sıçraması şudur: EF-G, ribozom üzerinde tRNA’ların ve mRNA’nın yer değiştirmesini sağlayan translasyonun motor proteinlerinden biridir. Fusidik asit, EF-G’nin döngüsünü bir noktada durduruyor; makineyi parçalamıyor, makineyi çalışırken kilitliyordu. Bu fikir, antibiyotik biyokimyasının en verimli fikirlerinden biridir: Bir sürecin “başlamasını” engellemek yerine, süreci “akarken” durdurmak, hücre için bazen daha yıkıcı olabilir.
Antibiyotik hikâyelerinde, keşifle birlikte direncin hikâyesi de başlar; çünkü antibiyotik kullanımı, bakteriler için güçlü bir seçilim baskısıdır. Fusidik asitte direnç, iki ana yoldan belirginleşir:
Bu dönem boyunca literatürde, klinik izolatlarda hangi mekanizmaların baskın olduğu, coğrafi dağılımlar ve kullanım pratikleriyle nasıl ilişkili bulunduğu gibi sorular giderek ağırlık kazanır. Topikal antibiyotik kullanımının yaygınlaşması, yerel direnç seçilimi açısından özellikle dikkat çekici bir eksen haline gelir; çünkü düşük ama süreğen ilaç maruziyeti, bakterilerin “kaçış” varyantlarını seçmede verimli bir ortam yaratabilir.
2000’li yıllar, translasyon makinesini atomik düzeyde görmenin mümkün hale geldiği dönemdir. X-ışını kristalografisi ve daha sonra hızla gelişen cryo-EM teknikleri, ribozom ve faktörlerinin “anlık fotoğraflarını” çıkarabilir hale gelir. Fusidik asit bu yeni çağda, yalnızca “etkili bir ilaç” olarak değil, aynı zamanda translasyonun nasıl işlediğini anlamak için bir “moleküler prob” olarak da değer kazanır.
2012’de yayımlanan önemli çalışmalar, fusidik asit direncinin EF-G üzerindeki mutasyonlarla nasıl ilişkili olabileceğini, bu mutasyonların bakteriye bazen “uygunluk bedeli” yüklediğini ve ikincil (telafi edici) mutasyonlarla bu bedelin nasıl azaltılabildiğini hem kinetik hem yapısal verilerle işler. Bu, antibiyotik direncini yalnızca bir “var/yok” niteliği olmaktan çıkarıp, evrimsel biyolojiyle iç içe geçmiş bir denge problemi olarak görmeyi kolaylaştırır: bakteri, direnç kazanırken büyüme gücünü kaybedebilir; ama doğa çoğu zaman bu kaybı telafi edecek yeni yollar bulur.
Aynı yıllarda FusB proteinlerinin yapısal ve işlevsel özellikleri de çözülür: FusB’nin EF-G’ye nasıl bağlandığı, ribozom döngüsünde hangi adımları “kurtardığı” ve fusidik asidin kurduğu kilidi nasıl gevşettiği gibi sorular, artık molekül molekül açıklanabilir hale gelir.
2020’lere gelindiğinde, fusidik asit araştırması iki ana nedenle yeniden alevlenir. Birincisi, antibiyotik direnci küresel ölçekte stratejik bir sağlık tehdidi olarak daha yüksek sesle konuşulmaktadır. İkincisi, cryo-EM ve tek molekül yaklaşımı gibi teknikler, yalnızca “yapıyı” değil, yapının zaman içindeki dönüşümünü yakalamaya yaklaşmıştır.
2020’de yayımlanan çalışmalar, FusB aracılı direncin yalnızca statik bir bağlanma olayı değil, EF-G’nin dinamiklerini ve ribozomla etkileşim ağını yeniden düzenleyen bir süreç olduğunu güçlü biçimde ortaya koyar. Bu, farmakoloji açısından kritik bir mesaj taşır: Direnç çoğu zaman “ilacı itmek” değildir; bazen ilacın kilitlediği makineyi, başka bir proteinin yardımıyla yeniden çalıştırmaktır.
2024–2025 döneminde ise bu yaklaşım daha da rafine hale gelir. Klinik izolatlarda fusidik asit direncinin genetik çeşitliliğini haritalayan çalışmalar, fusA mutasyonları ile fusB/fusC gibi genlerin birlikte nasıl görülebildiğini ve MRSA bağlamında hangi soyların öne çıktığını daha ayrıntılı biçimde betimler. Ardından 2025’te yayımlanan zaman çözünürlüklü cryo-EM temelli çalışmalar, FusB’nin fusidik asit tarafından ribozom üzerinde “tutuklanan” EF-G’ye bağlanarak, EF-G’de geniş ölçekli konformasyonel değişimler tetiklediğini ve böylece EF-G’nin ribozomdan ayrılmasının yeniden mümkün hale geldiğini mekanik bir anlatıya dönüştürür. Bu, yıllarca biriken biyokimyasal sezgilerin, artık “görülebilir” bir koreografiye dönüşmesidir.
Bugün fusidik asit, pek çok sağlık sisteminde özellikle cilt ve yumuşak doku enfeksiyonları bağlamında değerini korur; bazı ülkelerde sistemik kullanım deneyimi daha belirgindir ve belirli durumlarda (örneğin komplike stafilokokal enfeksiyonlar) kombinasyon stratejileri tartışılır. Bununla birlikte çağdaş yaklaşım, iki uyarıyı sürekli merkezde tutar:
Çağdaş araştırma ise klinik pratikle eşzamanlı iki hatta ilerler. Bir hat, direncin moleküler mekanizmalarını daha da ayrıntılandırarak yeni hedefler önermeye çalışır; diğer hat, fusidik asit iskeletinin kimyasal “oyun alanını” tarayarak daha iyi farmakokinetik, daha yüksek direnç bariyeri veya farklı spektrum özellikleri gösterebilecek analoglar tasarlamayı dener. 2025’te fusidik asit iskeletinin yapı–etki ilişkisini sistematik olarak genişleten çalışmalar, bu “yeniden tasarım” hattının canlılığını gösterir; 2024’te hesaplamalı kimya temelli türev simülasyonları ise modern antibiyotik geliştirmede in silico yaklaşımın artık ikincil değil, kurucu bir rol oynadığını hatırlatır.
Böylece fusidik asidin keşif öyküsü, bir toprak örneğinden başlayıp, ribozom üzerinde donmuş bir EF-G anına; oradan da zaman çözünürlüklü yapısal biyolojiyle izlenebilen direnç koreografisine uzanır. Bu hikâye, antibiyotik araştırmasının özünü de açık eder: keşif yalnızca yeni bir molekül bulmak değil, o molekülün biyolojide hangi düğümü tuttuğunu anlamak; ardından doğanın karşı hamlesini—direnci—çözerek yeni hamleler geliştirmektir.

Sinonim: Meticillin, methicillin
Metisilin, penisilinin yarı-sentetik analogu olan, beta-laktam sınıfı bir antibiyotiktir.
Eski adı Methicillin olan bu ilacın adı 2005 yılında meticillin olarak değiştirilmiştir. İlaç piyasasında artık üretilmeyen bu penisilin türevi ilacın yerini, gene penisilin türevi olan ve Meticilline benzeyen Oxacillin, Flucloxacillin ve Dicloxacillin adlı ilaçlar almıştır.
Gram pozitif bakterilerin sebep olduğu hastalıkları iyileştirmede kullanılır. Gram negatif bakteriler penisillin- resistent oldukları ilacın bu ilaçla tedavi edilemezler.
Meticillin-Resistent-Staphylococcus Aureus (MRSA) olarak bahsi geçen Staphylokok cinsi aslında sadece Meticillin’e karşı değil; penisiline karşı dirençli olduğu için penisilin türevleriyle tedavi edilemez.

Sinonim: cardiaca
Kalp hastalıklarına karşı kullanılan ilaçların tıbbi ismidir. Latincede, kalp anlamına gelen kardia kelimesinden gelir.
Başlıca iki grupta incelenebilir:
En önemli yan etkileri, yanlış kullanımlarında daha fazla kalp-ritm sorunlarına sebep olabilir; 2-3 kat fazla doz kullanımı direk ölümle sonuçlanabilir.
Sinonim: sedativa (çoğul)
Latincedeki, sedare kelimesinden gelir.
Hareketliliği azaltıcı, sakinleştirici bir psikotik ilaçtır. Korku yenici ya da çözücü olarak kullanılan sakinleştirici ilaçlarla karıştırılMAMALIdır.
Bu ilaç uyku getirici olarak da etki gösterir, fakat yüksek dozları bilincin kapanmasına sebep olabilir.
Ameliyatlardan önce bazı vakalarda stresi azalma amaçlı kullanılır.


1. Giriş ve Tarihçe
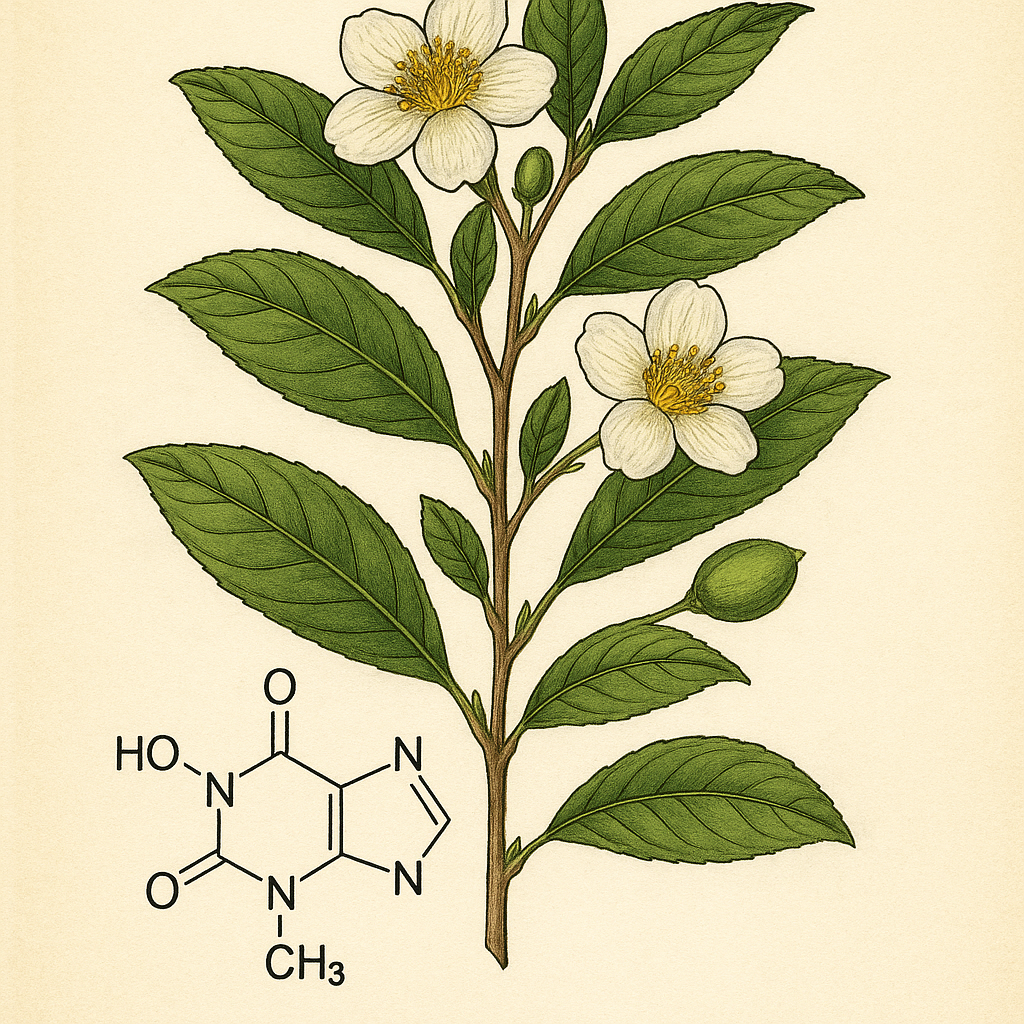
Teofilin (1,3-dimetilksantin), pürin bazlarından türetilmiş, ksantin yapısında bir bileşiktir. İnsan dokularında doğal olarak bulunmasının yanı sıra, bazı bitkilerde, özellikle de çay bitkisinde (Camellia sinensis) yüksek oranlarda saptanır. Teofilin ilk kez 1888 yılında Alman biyokimyacı Albrecht Kossel tarafından çay yapraklarından izole edilmiştir. İsmini çayın botanik cins adı olan Thea’dan alır. Farmakolojik etkileri 20. yüzyıl başlarında keşfedilmiş ve solunum yolu hastalıklarında bronkodilatör olarak kullanımı yaygınlaşmıştır.
2. Etki Mekanizması
Teofilinin etkileri çok yönlüdür ve birkaç biyokimyasal yolağa aynı anda etki eder. Bu etkiler başlıca solunum sistemi, kardiyovasküler sistem ve merkezi sinir sistemi üzerinde görülür.
2.1 Adenozin Reseptör Antagonizması
Teofilin, adenozin A1 ve A2 reseptörlerinin rekabetçi antagonisti olarak görev yapar. Adenozin normalde bronkokonstriksiyon, mast hücrelerinden histamin salınımı ve inflamatuar mediatörlerin aktivasyonu gibi süreçlerde rol oynar. Teofilin bu reseptörleri bloke ederek:
2.2 Fosfodiesteraz (PDE) İnhibisyonu
Teofilin, özellikle PDE3 ve PDE4 enzimlerini inhibe eder. Bu enzimler, hücre içi sinyal moleküllerinden biri olan cAMP’yi yıkar. Teofilin bu yıkımı inhibe ederek:
2.3 Kalsiyum Mobilizasyonu
Yüksek konsantrasyonlarda teofilin, sarkoplazmik retikulumdan kalsiyum salınımını artırarak kas hücrelerinin kasılma-gevşeme döngüsünü etkiler. Bu durum kalp kası hücrelerinde pozitif inotropik etki yaratabilir.
2.4 Anti-enflamatuar Özellikler
Teofilin, T lenfositler, eozinofiller ve nötrofiller gibi immün hücrelerde sitokin salınımını azaltır. Özellikle PDE4 inhibisyonu ile ilişkili olan bu etki, KOAH ve astım gibi hastalıklarda kronik hava yolu enflamasyonunun baskılanmasında önemli rol oynar.
3. Klinik Kullanım Alanları
3.1 Astım ve KOAH
Teofilin, özellikle uzun etkili bronkodilatörler ve inhale kortikosteroidlerle yeterli kontrol sağlanamayan olgularda ek tedavi olarak kullanılır. Bronşiyal düz kaslardaki gevşetici etkisiyle solunum yollarını açar.
3.2 Kardiyovasküler Sistem Üzerindeki Etkiler
3.3 Santral Sinir Sistemi ve Diğer Etkiler
4. Farmakokinetik Özellikler
5. Ticari Formülasyonlar
Bu ticari preparatlar farklı dozaj formlarında (oral tablet, IV form, SR tablet) bulunur ve hastanın klinik durumuna göre bireyselleştirilmiş doz rejimleri gerektirir.
6. Yan Etkiler ve Toksisite
Teofilin tedavisinde terapötik aralık dardır (5–15 µg/mL); bu nedenle plazma düzeylerinin dikkatli izlenmesi gerekir.
Teofilin, 19. yüzyılın sonlarında çay yapraklarından izole edilen doğal bir ksantin türevidir. Bu bileşik ilk kez 1888 yılında Alman biyokimyacı Albrecht Kossel tarafından tanımlanmıştır. Kossel, çay bitkisinden (Thea sinensis, günümüzde Camellia sinensis olarak adlandırılır) elde ettiği maddeleri incelerken bu yeni bileşiği keşfetmiş ve ona bitkinin cins adından türetilerek “teofilin” adını vermiştir. Bu keşif, o dönemde nükleik asitlerin ve pürin bazlarının yapısal analizine yönelik öncü çalışmalar arasında yer almaktadır. Ancak farmakolojik etkileri 20. yüzyılın başlarına kadar ayrıntılı olarak tanımlanmamış, özellikle bronkodilatör özellikleri 1920’li yıllarda araştırmalarla ortaya konmuştur.
Teofilinin sentez yöntemleri ilk kez 1895’te Emil Fischer ve Joseph von Mering tarafından geliştirilmiş, 20. yüzyılın başında ise bronşiyal astım tedavisinde klinik kullanımı başlamıştır. Teofilin zamanla kafein ve teobromin gibi diğer metilksantinlerle birlikte sınıflandırılmış ve özellikle solunum sistemi hastalıklarında önemli bir terapötik ajan olarak yerini almıştır.

“Digitalis” adı Latince digitus (parmak) kökünden gelir ve çiçeklerin parmaklık/fingerhut benzeri biçimine gönderme yapar; tür sıfatı purpurea sık görülen mor renge işaret eder. “Digitoksin” adı, Digitalis kökenini ve kardenolid sınıfı “-toksin” (burada toksisite vurgusu değil, kardenolitik glikozit üyeliğine tarihsel bir adlandırma) yapısını yansıtır.
Digitalis’in tıbbi kullanımı, 18. yüzyılda William Withering’in ödem ve kalp yetersizliği üzerindeki etkileri sistematik biçimde betimlemesiyle klinik farmakolojiye yerleşmiştir.
Digitoksin başlıca Digitalis purpurea (yabani yüksükotu) yapraklarından elde edilir. Bitkideki toplam kardenolit içeriği; tür, vejetasyon dönemi, hasat zamanı, kurutma tekniği ve ekstraksiyon yöntemine göre geniş aralıkta değişir. Literatürde, yaklaşık 10 kg ham bitki materyalinden koşullara bağlı olarak yaklaşık 6 g düzeyinde digitoksin fraksiyonu ayıklanabildiğine dair raporlar mevcuttur; bu değer standart değildir ve fitokimyasal değişkenliğe son derece duyarlıdır.
Digitoksin, Na⁺/K⁺-ATPaz pompasını hücre dışı bağlanma cebinden reversibl inhibe eder.
Bu etkiler, dolum basıncı yüksek ve sistolik fonksiyonu zayıf olgularda semptomatik iyileşmeye ve egzersiz toleransında artışa katkı sağlar; ancak dar terapötik aralık toksisite riskini beraberinde getirir.
Kılavuzlar Digitoksin/Digoksin’i birebir birincil seçenek olarak değil, kişiselleştirilmiş bir algoritmanın parçası olarak ele alır; ilaç seçimi eşlik eden komorbiditelere ve organ fonksiyonlarına göre yapılır.
Digitoksinin pazardaki mevcudiyeti bölge ve zamana göre değişkenlik gösterir. Tarihsel olarak oral tablet ve enjektabl formları bulunmuş olsa da, bazı ülkelerde üretici çekilmeleri ve tedarik kesintileri nedeniyle kesintili veya kısıtlı bulunabilir. Yerel güncel durumu doğrulamak gerekir. “Dünya genelinde hiç oral/parenteral form yoktur” biçimindeki kategorik ifadeler doğru değildir; bölgesel bağlam önemlidir.
Kardenolidler, bitkilerin otçul baskısına karşı geliştirdiği savunma metabolitleridir. Bazı kelebekler ve böcek hatları (ör. Danainae) Na⁺/K⁺-ATPaz’da hedef-yer değişimleri geliştirmiş, kardenolidleri hem tolere edip hem de vücutlarında biriktirerek kimyasal kamuflaj/savunma amacıyla kullanır. Bu durum, bitki-herbivor etkileşimlerinde klasik bir eş-evrim örneği olarak kabul edilir.
Yüksükotunun mor çiçekleri, Ortaçağ’da şifalı ot toplamayı bir tür hüner sayan köylü bilgeliklerinin arasında dolaşırdı; ama asıl sahneye çıkışı 18. yüzyılın sonlarında, Birminghamlı hekim William Withering’in sistematik gözlemleriyle olur. Withering’in titiz notları, “dropsy” diye anılan ödemli hastalarda Digitalis yapraklarının tuhaf ama tekrarlanabilir bir etkisi olduğunu söyler: nabız yavaşlar, idrar artar, nefes darlığı hafifler. İşin büyüsü, bitkinin “iyi geldiği” söylencesinden çok, tekrarlanabilirlik ve doz-ayarlaması fikridir; eczacılık ile klinik arasında bir köprü kurulur ve kalp glikozitlerinin uzun hikâyesi fiilen başlar.
19. yüzyılın ortalarında, eczacıların elinde imbikler, çözücüler ve kristaller vardır; ama bitkinin “hakiki” etkili maddesi bir türlü tek başına yakalanamaz. 1869’da Fransız eczacı Claude-Adolphe Nativelle, yüksükotundan yüksek derecede arıtılmış bir fraksiyonu “digitalin” adıyla duyurur. Bu “digitalin” aslında tek bir bileşik değil, kardenolid adı verilen etkili maddelerin bir karışımıdır; yine de o güne dek görülmemiş bir standartlaşma ufku açar. Nativelle’in şişeleri eczanelere girdiğinde, Digitalis artık yalnızca bir bitki değil, belli aralıklarla dozlanabilen bir ilaç ailesidir.
Bilim tarihi bazen tek bir laboratuvarın belirleyici hamlesini bekler. 1875’e gelindiğinde Strasbourg’daki genç farmakolog Oswald Schmiedeberg, digitalin sisinden sıyrılıp tek bir bileşiği—bugün bildiğimiz adıyla digitoksin—ayrı bir varlık olarak masaya koyar. Schmiedeberg’in yaptığı, bitkisel özütlerin tesadüfî faydasını kimyasal bireyselliğe tercüme etmektir; kâşiflik burada yalnız ayırma tekniğinde değil, “bir molekül – bir etki” fikrini cesurca savunmasındadır. Bu andan itibaren “kalp glikoziti” belirsiz bir şurup olmaktan çıkıp moleküler bir faile dönüşür.
İzleyen on yıllarda sahneye birkaç kilit isim daha girer. Freiburg’da kimyager Hermann Kiliani, glikozitlerin şeker kısmını aydınlatan yöntemler geliştirir; bugün eğitim kitaplarında “Keller–Kiliani reaksiyonu” diye anılan renk testleri, kardenolidlerin analitiğini standartlaştırır. Sterol kimyasının dev ismi Adolf Windaus, 1920’lerin ortasında glikozitlerin steroidal çekirdeğinin mimarisini çözmeye katkı verir; kardenolid halkasının (beş üyeli lakton) detayları ve halkaya bağlı şeker zincirlerinin (digitoksozlar) bağlanış biçimi böylece yavaş yavaş belirir. Kimyasal yapı netleştikçe, klinik gözlemlerdeki “pozitif inotropi, negatif kronotropi ve dromotropi” gibi kavramlar da Na⁺/K⁺-ATPaz üzerinden anlaşılabilir bir fiziolojiye bağlanır.
1930’larda sahneye bu ailenin bir başka üyesi, digoksin, Burroughs Wellcome araştırmacısı Sydney Smith’in Digitalis lanata’dan izolasyonu ile girer. Digoksin ile digitoksinin kaderleri bundan sonra sürekli karşılaştırılır: Biri daha hidrofilik ve renal yolla daha hızlı atılır; diğeri daha lipofilik, yüksek protein bağlanımlı ve uzun yarı ömürlüdür. Klinik, bu iki kardeşi farklı hasta tiplerinde farklı nedenlerle tercih etmeyi öğrenir; farmakoloji derslerinde ise öğrenciler, “aynı kapıyı (Na⁺/K⁺-ATPaz) çalan ama farmakokinetik kaderleri farklı iki ilaç” metaforuyla büyür.
Savaşlar biter, laboratuvarlar yeniden kurulur. 1940’ların sonu ve 1950’ler, standardizasyon ve biyoyararlanım tartışmalarının olgunlaştığı bir dönemdir. Radyoimmünoassay gibi ölçüm teknikleri doğmadan önce bile klinisyenler, dar terapötik aralıkla yaşamanın inceliklerini öğrenir: EKG’deki “digitalis etkileri”, hastanın iştahsızlığı, xantopsi anlatıları, elektrolitlerin (özellikle potasyum ve magnezyum) hayati rolü… 1960’larda antikor-temelli ölçümler ve Fab fragmanları (özellikle digoksin için) sahneye çıktığında, toplum artık bu ilaçların hem yaşam kurtaran, hem de ince ayar gerektiren karakterini içselleştirmiştir.
Bilimsel anlatılarda “durgunluk” pek nadir görülür; 20. yüzyılın son çeyreği ve 21. yüzyılın başı, iyon pompalarının yalnızca “pompalar” olmadığını, aynı zamanda sinyal platformları olduğunu gösterir. Na⁺/K⁺-ATPaz’ın Src ailesi kinazlarla ve NF-κB gibi yolaklarla kurduğu ilişkiler, kalp glikozitlerinin biyolojisini düz bir “pozitif inotropi” şemasının dışına taşır. Bu perspektiften bakınca digitoksin, kardiyak bir “mekanik anahtar” olmanın ötesinde, hücresel kaderi (apoptoz, proliferasyon, inflamasyon) belirleyen bir sinyal modülatörü gibi de görünür. Böylece laboratuvar defterlerine yeni başlıklar eklenir: onkojenik sinyal yolları, hipoksi tepkileri (HIF-1α), DNA hasar yanıtı, hatta viral replikasyon basamakları… Kardiyak glikozitlerin antitümör ve antiviral etkilere dair in vitro ve erken in vivo verileri, bir asırlık kalp ilacını ilaç yeniden konumlandırma literatürünün içine çeker.
Klinik sahada ise kalın çizgilerle yazılan gerçek şudur: digitoksin uzun yarı ömürlüdür (yaklaşık bir hafta), yüksek protein bağlanır, enterohepatik dolaşıma uğrar; bu yüzden doz değişiklikleri gecikmeli sonuç verir ve izlem sabır ister. Tam da bu nedenle, böbrek yetmezliği olan hastalarda (renal klirens ağırlıklı digoksine kıyasla) kimi zaman farmakokinetik avantaj sunabilir; ama karaciğer iş yükü ve etkileşimler bu tabloya yeni denklemler ekler. Klinik özelleşir, kılavuz algoritmaları kişiselleşir; kimi ülkelerde tedarik zincirleri digitoksini bir süre gölgeye iter, kimi merkezlerde ise deneyimli ekipler onunla çalışmayı sürdürür.
Ve bu hikâyenin güncel sayfaları hâlâ yazılıyor. HFrEF zemininde, modern kılavuz tedavilerine eklenen digitoksinin mortalite ve hastaneye yatış bileşik sonlanımları üzerindeki etkisini değerlendiren büyük çalışmalar, 2020’lerin ortasında yeniden gözlerin bu moleküle çevrilmesine yol açtı. Klinik istatistiklerin kurak cümleleri, aslında eski bir kahramanın sahneye güncellenmiş bir rolle dönebileceğini ima eder. Paralel kolda, kanser biyolojisi laboratuvarlarında NF-κB baskılanması, mitokondriyel yolların tetiklenmesi ve reaktif oksijen türlerinin yönetimi üzerinden digitoksinin antitümör etkileri irdelenir; viroloji masalarında ise koronavirüsler başta olmak üzere, çeşitli solunum yolu virüslerinde kalp glikozitlerinin giriş-sonrası basamaklara etki eden geniş spektrumlu inhibitörler olabileceğine dair veriler birikir. Bütün bunlar klinik pratiğe “yarın sabah” değil belki, ama dikkatli tasarlanmış denemeler ve biyobelirteç odaklı seçilmiş hasta grupları eşliğinde girebilecek sinyallerdir.
Elbette bir bilim hikâyesi, kâşifler olmadan eksik kalır. Bu yolculukta Withering taşları döşedi; Nativelle karışımı damıttı; Schmiedeberg tek bir molekülü sahneye çıkardı ve “digitoksin” kimliğe kavuştu. Kiliani, şekerlerin sırlarını açtı; Windaus, sterol çekirdeğinin mimarisini yerleştirdi. Sydney Smith, kardeş bileşik digoksini izole ederek aileyi genişletti. Klinik cephesinde James Mackenzie, atriyal fibrilasyonu tanımlayarak hedefe ışık tuttu; Arthur Robertson Cushny, farmakolojik etkilerin fiziolojik temellerini çizdi. Kimya ile klinik, eczane ile koğuş, bitki ile pompa arasında gidip gelen bu uzun diyaloğun adı digitoksin oldu.
Bugün yüksükotu tarlasına bakan biri, mor haznenin içine eğilmiş arıların telaşını görür; farmakoloğun baktığı yerde ise Na⁺/K⁺-ATPazın dış yüzeyinde bekleyen bir kardenolid silueti vardır. O siluet, iki yüzyıldır kalp atımlarının ritmini değiştirmekle kalmaz; hücrenin tepkilerini, sinyallerini ve kaderini de bir tutam kimya ile yeniden yazar. Digitoksinin keşif tarihini “tamamlanmış” sanmak, bu yüzden, bu hikâyenin doğasına aykırıdır: keşif burada bir olay değil, süren bir süreçtir; bitki, molekül ve insan arasında yazılmaya devam eden bir süreç.

Ticari isimler; Digoxin Sandoz®, Juvisé®, Lanitop
“Digoksin” terimi, yüksük otu bitkisinin cins adı olan “Digitalis”ten ve farmakolojide doğal kaynaklardan türetilen maddeleri belirtmek için sıklıkla kullanılan “-in” son ekinden türetilmiştir. “Digi-” öneki, gelişiminin temel taşı olan bitkiye saygı duruşunda bulunurken, “-oxin” ise bitkinin organik kimyasal doğasını ifade eder.
Digoksin, yüksük otu bitkisinden elde edilen, kalp yetmezliği ve kardiyak aritmiler gibi çeşitli kalp rahatsızlıklarının tedavisinde yaygın olarak kullanılan güçlü bir kardiyak glikozittir. Tedavi aralığının dar olması ve yeni ilaçların bulunması nedeniyle kullanımı yıllar içinde azalmış olsa da tıbbi ilgi konusu olmaya devam ediyor. Bu makale Digoksin’in farmakolojik yönlerini, klinik endikasyonlarını ve güvenlik hususlarını ele almaktadır.

Digoksin, diğer bir kalp glikozidi olan Digitoksin’den, 12. karbon atomunda bir hidrojen molekülüne sahip olmasıyla ayrılır. Bu yapısal varyasyon farmakokinetiğini ve farmakodinamiğini etkiler.
Digoksinin temel etki mekanizması, kalp kası hücrelerinde Na+/K+-ATPaz’ın inhibisyonunu içerir. Bu inhibisyon, Na+ ve Ca2+ değişimini bozar, hücre içi Ca2+ konsantrasyonunun artmasına neden olur, bu da kalp kası kasılmalarının gücünü ve hızını artırır. Ek olarak ilacın baroreseptörlerin duyarlılığını arttırdığı ve nörohormonal etkilerini de açıkladığı düşünülmektedir.
Digoksin, aşağıda belirtildiği gibi kalp kası fonksiyonu üzerinde birkaç önemli etki gösterir:
Sonuç olarak digoksin atım hacmini iyileştirir, böbreklere kan akışını artırır ve idrar çıkışının artmasına yol açar. Ancak bu pozitif inotropik etkiler yalnızca sağlıklı kalp kası liflerinde görülür.
Digoksin öncelikle akut ve kronik kalp yetmezliğinin yanı sıra atriyal fibrilasyon ve çarpıntının tedavisinde kullanılır. Dozaj ürün yönergelerine göre yapılır ve ilacın dar terapötik aralığı nedeniyle yakın takip gerektirir. Böbrek fonksiyon bozukluğu olan hastalarda doz ayarlaması gereklidir.
Digoksin, aşırı duyarlılık, dijital zehirlenmesi, bazı kardiyak aritmiler gibi çeşitli durumlarda ve hipokalemi, hiperkalsemi ve hipomagnezemi gibi elektrolit dengesizliklerinin varlığında kontrendikedir.
İlaç, P-glikoproteinin bir substratıdır ve bu taşıyıcının inhibitörleri, digoksin serum seviyelerini yükseltebilir. Glikozit toksisitesinin artması nedeniyle kalsiyumun digoksinle birlikte intravenöz olarak uygulanmaması gerekir. Beta blokerler, kalsiyum kanal blokerleri ve diüretikler gibi bazı ilaçlar digoksinle etkileşime girebilir ve dikkatli takip gerektirir.
Hem Digoksin hem de Eliquis, P-glikoprotein taşıma sisteminin substratlarıdır. Bu nedenle, bunların eş zamanlı alınması her ilacın düzeyini etkileyebilir ve dikkatli izlemeyi gerektirir. Digoksin seviyelerindeki bir artış digoksin toksisitesine yol açabilirken, Eliquis’in yüksek seviyeleri aşırı kanamaya neden olabilir.
Böbrek yetmezliği
Her iki ilaç da kısmen böbrekler yoluyla atılır. Böbrek yetmezliği olan hastalarda doz ayarlaması ve böbrek fonksiyonunun izlenmesi gerekebilir.
Hemodinamik Etkiler
Digoksinin pozitif inotropik etkileri, Eliquis gibi kalp kasılması üzerinde herhangi bir yararlı etki göstermeyen ancak pıhtı oluşumunu önlemek için gerekli olan antikoagülanlar tarafından bir şekilde dengelenebilir. Hastanın klinik durumuna bağlı olarak ilaçlardan birinin veya her ikisinin dozajının ayarlanmasına ihtiyaç duyulabilir.
Polifarmasi
Digoksin ve antikoagülanlar arasındaki etkileşim diğer ilaçlardan etkilenebileceğinden, diğer antiaritmikler, diüretikler veya antitrombosit ajanlar da dahil olmak üzere birden fazla ilaç kullanan hastaların özellikle dikkatli olmaları gerekir.
Klinik Kılavuzlar
Hem digoksin hem de antikoagülan reçete edilen hastalar için ilaç seviyelerinin, böbrek fonksiyonunun ve pıhtılaşma parametrelerinin rutin olarak izlenmesi şiddetle tavsiye edilir. Her iki ilaç sınıfının da dar terapötik pencereleri vardır ve etkileşimler kanama veya digoksin toksisitesi gibi olumsuz etkilere yol açabilir.
Dar terapötik penceresi nedeniyle digoksin sıklıkla kardiyak aritmiler, bulantı, kusma ve yorgunluk gibi olumsuz etkilere yol açar. Nadir durumlarda algı bozuklukları ve aşırı duyarlılık reaksiyonları gibi daha ciddi yan etkiler gözlemlenmiştir.
Digoksin, kalp hastalıklarının tedavisinde hayati ancak karmaşık bir araç olmaya devam ediyor. Kullanımı, farmakolojik profilinin, ilaç etkileşim potansiyelinin ve yakın terapötik izleme gerekliliğinin kapsamlı bir şekilde anlaşılmasını gerektirir.
Digoksin, yüzyıllardır bilinen botanik bir tür olan yüksük otu bitkisinin (Digitalis purpurea) yapraklarından elde edilir. Yüksük otu’nun tıbbi kullanımı, 18. yüzyılda, etkilerini titizlikle belgeleyen ve 1785 tarihli “An Account of the Foxglove” adlı kitabında dozları tavsiye eden İngiliz doktor William Withering tarafından popüler hale getirildi.

19. ve 20. yüzyıllar boyunca yüksük otu bitkisinin aktif bileşikleri izole edildi ve sentezlendi; bu da digoksin ve dijitoksin gibi kardiyak glikozitlerin gelişmesine yol açtı. Digoksin, 1930’larda klinik uygulamaya girdi ve kalp tedavisi için daha istikrarlı ve etkili bir seçenek sundu.
Zaman İçinde Farmakolojik Etki
Digoksin 20. yüzyıl boyunca yaygın klinik kullanıma kavuştu. Kalp kası kasılma kuvvetini artıran güçlü inotropik etkileri nedeniyle kalp yetmezliği ve atriyal fibrilasyon gibi durumların tedavisinin temel dayanağı haline geldi. Ancak yeni ilaçların artması ve digoksinin dar terapötik penceresinin anlaşılması, son yıllarda kalp yetmezliğinde kullanımının azalmasına yol açmıştır.

Flukonazol, triazol antifungaller grubuna ait, sistemik ve mukozal mantar enfeksiyonlarının tedavisi ve profilaksisinde yaygın kullanılan bir antifungal ilaçtır. Dünyada en bilinen ticari ismi Diflucan® olmakla birlikte pek çok jenerik preparat da mevcuttur. İlacın yapısal özellikleri ve farmakokinetik profili, onu özellikle Candida türlerine ve Cryptococcus neoformans’a bağlı enfeksiyonlarda bir “temel ilaç” konumuna yerleştirmiştir; bu nedenle Dünya Sağlık Örgütü’nün Temel İlaçlar Listesi’nde de yer almaktadır.
Flukonazolün klinik önemini artıran başlıca özellikleri şunlardır:
Flukonazol, Pfizer tarafından 1980’li yılların başında geliştirilmiş, 1981’de patentlenmiş ve 1988 civarında klinik kullanıma girmiştir. ABD’de FDA tarafından onayı 29 Ocak 1990 tarihinde verilmiştir.
Adlandırma (etimoloji):
Böylece fluconazole, yaklaşık olarak “flor içeren konazol türevi antifungal” anlamına gelir. “Konazol” son eki, imidazol ve triazol çekirdeği içeren geniş bir antifungal sınıfını (ketokonazol, mikonazol, itrakonazol, vorikonazol vb.) işaret eder; bu isimlendirme, farmakolojik sınıfın hızlıca tanınmasını sağlayan tipik bir INN (International Nonproprietary Name) örneğidir.
Molekül, iki adet 1,2,4-triazol halkası ve bir diflorofenil çekirdek etrafında örgütlenen simetrik bir yapıya sahiptir. Bu yapı:
sağlayarak, hem sistemik enfeksiyonlara ulaşacak doku penetrasyonunu, hem de dozlama kolaylığını mümkün kılar.
Sınıflandırma:
Flukonazol, azol grubunun tipik bir üyesi olarak ergosterol biyosentez yolunu hedef alır. Spesifik etki noktası:
Bu enzimin işlevi, lanosterolün 14α-demetilasyonu yoluyla ergosterol sentezini gerçekleştirmektir. Ergosterol:
için kritik bir sterol bileşenidir; mantar hücreleri için memeli hücrelerindeki kolesterol ile işlevsel açıdan yakın paralellik gösterir.
Flukonazolün etki mekanizması özetle:
Flukonazol çoğu Candida türü için fungistatik, bazı mantarlara (örneğin Cryptococcus spp.) karşı ise doz ve koşullara bağlı olarak fungisidal etki gösterebilir.
Seçici toksisite:
Bu iki özellik, flukonazolün mantarlara karşı seçici toksisitesinin temelini oluşturur. Bununla birlikte, insan karaciğerinde bazı sitokrom P450 izoenzimlerini (özellikle CYP2C19, kısmen CYP2C9 ve CYP3A4) inhibe etmesi, klinik düzeyde önemli ilaç etkileşimlerine yol açabilir.
Flukonazolün farmakokinetik profili, klinikte kullanımını son derece pratik kılan bir dizi avantaj sunar.
Bu özellikler, aynı dozun hem oral hem intravenöz yolla benzer sistemik maruziyet sağlamasına imkan verir ve pek çok durum için IV’den oral geçiş (switch) stratejisini kolaylaştırır.
Bu dağılım özellikleri, özellikle kriptokokal menenjit, kandideminin sekonder odakları, vajinal kandidiyaz ve deri/mukozal Candida enfeksiyonları için etkinlik açısından kritik önemdedir.
Bu durum:
ile ilişkilidir (yine de belirgin hepatik yetmezlikte dikkat gereklidir).
Böbrek yetmezliği: Kreatinin klerensine paralel olarak flukonazol klerensi de azalır; ağır böbrek yetmezliğinde yarı ömür 90 saate kadar uzayabilir. Bu nedenle doz azaltımı veya doz aralığının uzatılması gereklidir.
Flukonazol, çok sayıda mantar enfeksiyonunda hem tedavi hem de profilaksi amacıyla kullanılır. Klinik kılavuzlar ve ürün bilgileri temel alındığında başlıca endikasyonlar şu şekilde özetlenebilir:
Vajinal kandidiyaz için yetişkinlerde klasik rejim 150 mg tek doz oral flukonazoldür; rekürren olgularda aylık 150 mg profilaktik dozlar kullanılabilir.
Bu bağlamda flukonazol genellikle stabil, ağır olmayan tablolarda veya duyarlı türlerin söz konusu olduğu durumlarda, bazen ekinokandinlerden idame tedaviye geçiş amacıyla kullanılır.
Flukonazolün aktivite spektrumu, başta Candida ve Cryptococcus olmak üzere çeşitli mantarları kapsar; buna ek olarak bazı dimorfik mantarlara karşı da etkindir:
Bununla birlikte, Candida krusei (Pichia kudriavzevii) ve sıklıkla Candida glabrata (Nakaseomyces glabrata) gibi türler doğal veya kazanılmış flukonazol direnci nedeniyle, klinikte flukonazole genellikle dirençli kabul edilir.
Flukonazol, aşağıdaki yüksek riskli hasta gruplarında Candida enfeksiyonlarının profilaksisi için kullanılabilir:
Flukonazol; tablet/kapsül, oral süspansiyon ve intravenöz infüzyon çözeltisi formlarında mevcuttur. Doz, endikasyona, hastanın klinik durumuna, böbrek fonksiyonuna ve eşlik eden ilaçlara göre ayarlanır. Aşağıdaki aralıklar, sık kullanılan örnek rejimleri göstermektedir; gerçek klinik uygulama mutlaka güncel kılavuz ve uzman hekim değerlendirmesine dayanmalıdır.
Yetişkin örnek dozları (özet):
Çocuklarda doz: Genellikle mg/kg/gün üzerinden hesaplanır (örneğin 3–12 mg/kg/gün aralığında). Yenidoğan ve infantlarda doz aralığı ve sıklığı, yarı ömrün yaşa bağlı değişimi dikkate alınarak ayarlanır.
Böbrek yetmezliği:
Kreatinin klerensi azaldıkça idame dozu azaltılır (örneğin kreatinin klerensi <50 mL/dk ise doz %50’ye indirilebilir); hemodiyaliz seansları flukonazolü uzaklaştırdığı için diyaliz sonrası ek doz gerekebilir.
Mutlak kontrendikasyonlar:
Dikkat gerektiren durumlar:
Flukonazol genellikle iyi tolere edilir; yan etkilerin büyük kısmı hafif ve geçicidir. Bununla birlikte, nadir fakat ciddi advers etkiler de bildirilmiştir.
Sık (≥ %1):
Daha seyrek:
Nadir fakat ciddi:
Uzun süreli yüksek doz flukonazol kullanımı ile, gebeliğin ilk trimesteri sırasında özgül bazı konjenital malformasyon kümeleri bildirilmiştir; ayrıca daha düşük dozlarla dahi spontan abortus riskinde artış saptandığını gösteren epidemiyolojik veriler bulunmaktadır (mutlak risk yine de düşük kalmakla birlikte).
Flukonazol, insan sitokrom P450 sistemi üzerinde özellikle:
Bu nedenle aşağıdaki ilaçlarla önemli farmakokinetik etkileşimler görülebilir:
Farmakodinamik düzeyde, QT aralığının uzaması potansiyeli, eşzamanlı elektrolit bozuklukları veya yapısal kalp hastalıkları ile birleştiğinde klinik açıdan anlamlı hale gelebilir; bu durumlarda EKG ve elektrolit izlemi önerilir.
Flukonazol, uzun yıllar boyunca geniş kullanım görmüş, bu da özellikle Candida türleri arasında önemli ölçüde azole direnci gelişmesine yol açmıştır. Direnç, çoğu zaman kademeli olarak ve genellikle uzamış veya tekrarlayıcı tedaviler sırasında ortaya çıkar.
Flukonazol direncinin önde gelen mekanizmaları şunlardır:
Evrimsel düzeyde bakıldığında, insan tarafından yaygın ve kronik azol kullanımı:
Bu süreçte:
Klinik pratikte bu, özellikle yüksek riskli hastalarda:
gibi stratejilerin önemini artırmaktadır.
Epidemiyolojik çalışmalar:
Bu nedenle:
Flukonazol, anne sütüne plazma düzeylerine yakın konsantrasyonlarda geçer. Kısa süreli düşük doz tedavilerde emzirme çoğunlukla mümkün kabul edilse de, uzun süreli yüksek doz rejimlerde, klinik durum özelinde değerlendirme yapılmalıdır.
Flukonazol, neonatal dönem de dahil olmak üzere çocuklarda kullanılabilir; ancak farmakokinetik parametreler yaşa göre değişir:
Dozlar genellikle mg/kg/gün üzerinden titizlikle hesaplanmalı ve özellikle böbrek fonksiyonları dikkate alınmalıdır.
Flukonazol, günümüzde azol spektrumunun “ilk kuşak” sistemik triazollerinden biri olarak düşünülür. İleri kuşak azollerle karşılaştırıldığında:
Buna karşın, hafif–orta şiddette Candida enfeksiyonları, kriptokokal menenjitin idame tedavisi ve belirli profilaktik endikasyonlarda, flukonazol hâlâ küresel ölçekte klinik pratiğin temel taşı olmaya devam etmektedir.
Flukonazol sahneye çıkmadan önce sistemik mantar enfeksiyonlarının tedavisinde dünya neredeyse iki temel direğe dayanıyordu:
Bu dönemde yoğun bakım olanaklarının artması, kanser tedavisinde kemoterapi rejimlerinin agresifleşmesi ve organ nakillerinin yaygınlaşmasıyla, ciddi invazif mantar enfeksiyonları artık istisnai vakalar değil, klinik pratiğin giderek büyüyen bir parçası haline gelmişti. Aynı yıllarda, 1980’lerin başından itibaren HIV/AIDS pandemisi, özellikle kriptokokal menenjit ve uzamış kandidemi gibi enfeksiyonları, büyük şehir hastanelerinde günlük rutinin acı bir parçası haline getirdi.
Klinisyenler, damar yolunu harap etmeyen, böbreği çökertmeyen ama yine de sistemik olarak güçlü bir antifungal ajana ihtiyaç duyuyordu. İşte flukonazol araştırma programı, tam bu klinik baskının ve farmakolojik arayışın kesiştiği noktada doğdu.
Pfizer, 1970’lerin sonuna gelindiğinde İngiltere’nin Sandwich, Kent kentindeki araştırma laboratuvarlarında yeni bir antifungal tasarlamak üzere sistematik bir program başlattı. Bu programın resmi olarak 1978’de başladığı, daha sonra yayımlanan “Discovery of fluconazole, a novel antifungal agent” başlıklı makalede ayrıntılı biçimde aktarılır.
Programın liderlerinden biri, organik kimyacı Ken Richardson idi; Pfizer’in Sandwich’teki araştırma departmanında çalışan bu ekip, daha sonra flukonazol nedeniyle dünya çapında tanınacaktı. Ulusal Buluşçular Onur Listesi (National Inventors Hall of Fame) ve benzeri kaynaklarda, flukonazolün keşfi özellikle Ken Richardson’a atfedilir; ancak bu keşif, kolektif bir laboratuvar emeğinin ürünüdür.
Richardson’ın yanı sıra, erken ve temel katkılarıyla öne çıkan isimler arasında şunlar sayılır:
Bu araştırmacılar, Pfizer’in Sandwich araştırma biriminde, hem tasarım hem de in vivo modeller üzerinde çalışan çekirdek kimya–farmakoloji ekibini oluşturuyordu.
Programın başlangıç noktasında ekip, o dönem halihazırda bilinen azol antifungallerden hareket etti:
Ancak erken çalışmalar gösterdi ki:
Bunun üzerine program, adım adım:
Bu stratejinin amacı şuydu:
Sonuçta, bir dizi bis-triazol prototipi sentezlendi ve bunlar, sistemik ve yüzeyel mantar enfeksiyonlarının hayvan modellerinde test edildi.
Flukonazol, erken araştırma safhasında UK-49,858 kod adıyla anılıyordu. Bu bileşik:
Hayvan çalışmalarında UK-49,858:
Bu preklinik veri yığını, tedricen tek bir molekül üzerinde yoğunlaşılmasına yol açtı:
hepsini bir arada en iyi sağlayan yapı, bugün bildiğimiz adıyla flukonazol oldu.
1980’lerin ortasına gelindiğinde, UK-49,858 artık yalnızca bir laboratuvar kodu değil, insan klinik denemelerine geçmek üzere olan bir adaydı. Çeşitli faz II benzeri erken çalışmalar:
Bu erken klinik serilerde görülen ana temalar:
Bu veriler, 1988 civarında ticari kullanıma girişin ve kısa süre sonra da ABD FDA başvurusunun önünü açtı. Flukonazol, birçok kaynakta 1988’de klinik kullanıma giren, 1990’da ise FDA onayı alan bir molekül olarak tanımlanır.
ABD Gıda ve İlaç İdaresi (FDA), flukonazolu Diflucan® ticari adıyla 29 Ocak 1990 tarihinde onayladı.
Bu onayın çarpıcı noktalarından biri, onay sürecinin görece kısa olmasıdır:
Flukonazol, kısa sürede:
standart tedavi protokollerinin merkezine yerleşti.
Özellikle AIDS’li hastalarda:
flukonazol mortaliteyi anlamlı biçimde azaltan, günlük pratiği değiştiren bir ajan haline geldi.
Keşfin isimlendirilmesinde çoğu zaman Ken Richardson tek başına anılsa da, flukonazol araştırma programı kolektif bir takım çalışmasıydı. “Discovery of fluconazole, a novel antifungal agent” makalesinde yazarlar şöyle listelenir:
Bu isimler, Pfizer’in Sandwich, Kent’teki Araştırma Departmanı’na bağlıydı ve daha sonra yayımlanan birçok hayvan çalışmasının da yazar listelerinde tekrar tekrar karşımıza çıkar.
Böylece flukonazol, yalnızca “bir buluşçu”nun değil, klinik farmakoloji, sentetik kimya, farmakokinetik ve deneysel mikrobiyolojinin birlikte çalıştığı bir multidisipliner orkestranın ürünü olarak tarih sahnesine çıktı.
1990’ların başında AIDS pandemisi küresel olarak derinleşirken, kriptokokal menenjit özellikle Sahra-altı Afrika’da, bağışıklık sistemi baskılanmış HIV pozitif bireylerde önde gelen ölüm nedenlerinden biriydi. Flukonazol, bu bağlamda üç düzeyde önemli oldu:
Bu dönemde flukonazol, bir yandan zengin ülkelerde kanser hastalarında invazif kandidiyaz profilaksisinin parçası olurken, diğer yandan yoksul ülkelerde AIDS’le mücadelede bir “hayat çizgisi”ne dönüştü.
Flukonazolün popüler imajı, birkaç klinik özelliğin birleşmesiyle şekillendi:
Bu nedenle bazı klinisyenler ve hasta toplulukları arasında flukonazol, özellikle:
olarak anılır hale geldi (elbette bunlar resmi tıbbi terimler değil, pratiğin içindeki gayriresmi lakaplardır).
Bununla birlikte, rehberler giderek daha temkinli bir çizgiye kaydı; örneğin:
Yine de flukonazol, özellikle vajinal kandidiyaz tedavisinde obstetrik ve jinekoloji pratiğinde, günümüzde de en sık reçete edilen ajanlardan biri olmaya devam eder.
Flukonazol, zamanla sadece tedavi değil, profilaksi amaçlı da protokollere girdi:
Diğer yandan, daha heterojen bir grup olan:
için flukonazol profilaksisi, daha çok merkez ve vaka bazlı kararlarla yürütülmekte; burada odak, sadece klinik faydayı maksimize etmek değil, aynı zamanda direnç gelişimi riskini dengede tutmaktır.
Flukonazol ne kadar yaygın kullanıldıysa, ona karşı direnç evrimi de o kadar hızlandı.
2010’lu yıllar boyunca yapılan çalışmalar, Candida albicans’ın flukonazol karşısında:
gibi çok katmanlı mekanizmalarla direnç geliştirebildiğini gösterdi.
2019’da yayımlanan bir çalışma, flukonazol maruziyetinin ilaçla indüklenen çiftleşme yetkinliği ve paraseksüel rekombinasyon üzerinden, çoklu direnç mekanizmalarını aynı hücre soyu içinde birleştiren bir evrimsel yol sunduğunu göstererek bu tabloya daha da sofistike bir boyut ekledi.
Son yıllarda Candida parapsilosis’te flukonazol direncinin dünya çapında kümelenmiş salgınlar halinde ortaya çıktığı; bazı bölgelerde flukonazole direnç oranlarının dramatik şekilde arttığı gösterildi. Bu durum, cilt florasında kolonize olabilen ve kateterle ilişkili kandidemiye yol açabilen bu türü, yoğun bakım ve hematoloji üniteleri için kritik bir problem haline getirdi.
Son on yılın en çarpıcı mantar tehditlerinden biri, Candida auris (son bazı kaynaklarda yeni taksonomik adlandırmalarla anılsa da klinik pratikte hâlâ bu isim kullanılıyor) oldu.
Çok sayıda çalışma ve saha raporuna göre:
Bu tablo, flukonazolü bir zamanlar “geniş spektrumlu, pratik” antifungal yapan pek çok özelliğin, günümüzün dirençli patojen manzarasında artık kendiliğinden başarı garantisi sunmadığını gösteriyor.
Dirençli Candida suşlarına karşı tek başına flukonazolün gücünün azaldığı anlaşıldıkça, araştırmacılar kombinasyon tedavileri üzerinde yoğunlaştı.
Özellikle ilgi çeken hatlardan biri, flukonazolün bitkisel kökenli bileşiklerle kombinasyonu oldu:
Başka bir araştırma hattı ise, statinler ve diğer lipid metabolizması üzerinde etkili ilaçların, flukonazol ile birlikte kullanımında sterol biyosentez yolaklarında “iki noktalı darbe” (dual hit) yaratma potansiyeli üzerinde duruyor; bu bağlamda statinlerin yeniden konumlandırılması (“drug repurposing”) tartışılıyor.
Flukonazol bugün için yeni bir molekül değil; patentleri 2000’li yılların ortalarında büyük ölçüde sona erdi ve onlarca jenerik formu mevcut. Ancak farmasötik teknoloji, bu eski molekülü yeni şekillerde “yeniden keşfetmeye” devam ediyor:
Bu çalışmaların ortak paydası, flukonazolün artık “yeni” bir kimyasal varlık olmamasına rağmen, formülasyon bilimi sayesinde terapötik profilinin hâlâ rafine edilebilir olmasıdır.
2020’lerin ortasına gelindiğinde flukonazol, küresel reçete istatistiklerinde hâlâ en sık kullanılan antifungallerden biri; yalnızca ABD’de milyonlarca reçeteyle ilk 200 ilaç arasında yer alıyor.
Eşzamanlı olarak:
gösteren geniş bir literatür birikmiş durumda.
Güncel araştırma gündemi, bu nedenle flukonazol etrafında üç ana soruya odaklanmış durumda:
Flukonazolün hikâyesi, bu açıdan tamamlanmış bir geçmiş anlatısı değil; aksine, modern antifungal tedavinin evrimsel dinamikleriyle birlikte yazılmaya devam eden, hâlâ aktif bir klinik ve mikrobiyolojik öykü niteliği taşıyor.
Yorum yazabilmek için oturum açmalısınız.