
İçindekiler
1) Tanım
Galaktozemi, galaktozun metabolik yolaklarda glikoz metabolitlerine dönüştürülememesi sonucu galaktoz ve toksik türevlerinin birikimiyle seyreden, otozomal resesif geçişli nadir bir metabolizma hastalığıdır. Klinik tablo akut neonatal karaciğer yetmezliği ve sepsis tablosundan, uzun dönemde nörokognitif bozukluklar ve gonadal disfonksiyonlara kadar geniş bir spektrum gösterir.
2) Biyokimyasal Temel
Laktoz, bağırsaklarda laktaz enzimi ile glukoz ve galaktoza ayrılır. Galaktoz, hepatositlerde Leloir yolu üzerinden glukoz-1-fosfat ve UDP-glukoz metabolitlerine çevrilir. Bu dönüşüm için üç ana enzim kritik rol oynar: galaktokinaz (GALK1), galaktoz-1-fosfat üridil transferaz (GALT) ve UDP-galaktoz 4-epimeraz (GALE). Bu enzimlerden herhangi birindeki doğuştan eksiklik, galaktoz metabolizmasının farklı tip bozukluklarına yol açar.
3) Epidemiyoloji
Klasik galaktozeminin sıklığı yaklaşık 1/40.000 olarak bildirilmektedir. Ancak insidans etnik ve coğrafi farklılıklar gösterir. Bazı izole popülasyonlarda çok daha yüksek oranlara rastlanmıştır.
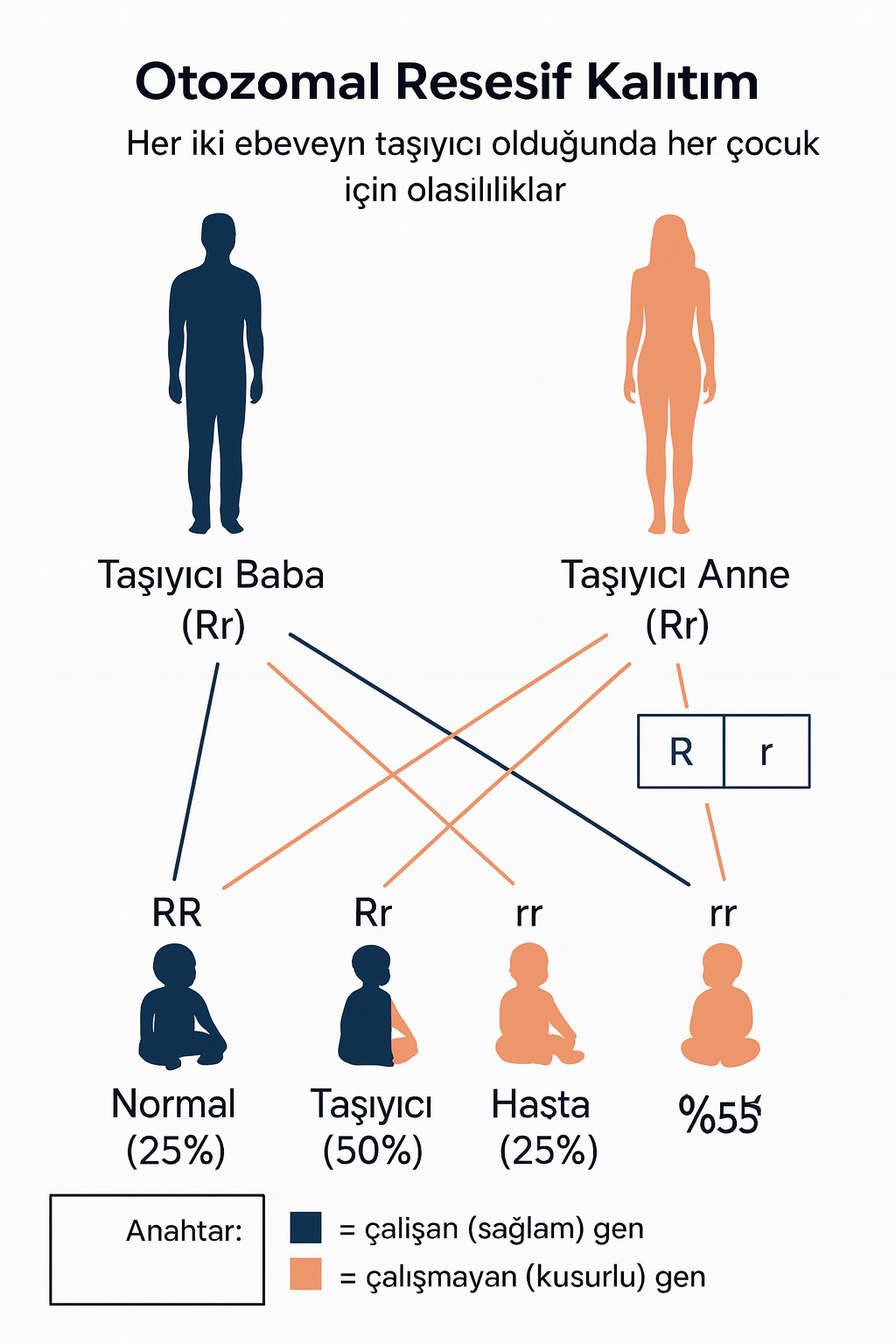
4) Genetik
Klasik galaktozemi, kromozom 9p13 üzerinde yer alan GALT genindeki mutasyonlarla ilişkilidir. En sık gözlenen mutasyonlar p.Q188R ve p.K285N’dir. Duarte (D2) varyantı ise p.N314D polimorfizmi ile ilişkili olup, genellikle klinik olarak hafif veya asemptomatiktir. Galaktokinaz eksikliği (GALK1 geni) ve UDP-galaktoz 4-epimeraz eksikliği (GALE geni) daha nadir alt tiplere yol açar.
5) Patofizyoloji
GALT enziminin eksikliği, galaktoz-1-fosfatın glukoz-1-fosfata dönüştürülememesine neden olur. Galaktoz-1-fosfat, galaktitol ve diğer metabolitlerin birikimi ATP tükenmesine, fosfatın tutulmasına, glikozilasyon bozukluklarına ve enzim inhibisyonlarına yol açar. Özellikle karaciğer, böbrek, beyin, göz merceği ve ovaryumlar etkilenir. Galaktitolün lenste birikimi katarakt oluşumuna neden olur.
6) Klinik Belirtiler
Hastalar genellikle doğumda sağlıklıdır. Belirtiler, süt alımının başlamasıyla birlikte yaşamın ilk günlerinde ortaya çıkar. Klinik tablo beslenme güçlüğü, kusma, ishal, kilo kaybı, hipotoni, hepatosplenomegali, sarılık, koagülopati, hipoglisemi, letarji ve konvülziyonları içerir. Escherichia coli sepsisi ve menenjiti sık görülür. Ayrıca birkaç hafta içinde katarakt gelişebilir. Tedavi edilmeyen olgularda mortalite yüksektir.
7) Uzun Dönem Sonuçlar
Diyet tedavisine rağmen hastalarda kalıcı sorunlar görülebilir. Bunlar arasında nörogelişimsel gerilik, konuşma apraksisi, ince motor beceri bozuklukları ve tremor yer alır. Kadınlarda prematür ovaryan yetmezlik sık görülürken erkeklerde genellikle normal puberte ve fertilite gözlenir.
8) Sınıflandırma
- Tip I (klasik galaktozemi): GALT eksikliği.
- Tip II: Galaktokinaz eksikliği. Klinik olarak genellikle infantil katarakt ile sınırlıdır.
- Tip III: UDP-galaktoz 4-epimeraz eksikliği. Periferik form asemptomatik olabilir, genelleşmiş form klasik tabloya benzer ağır semptomlarla seyreder.
9) Tanı
Yenidoğan taraması ile üçüncü günden itibaren tanı konabilir. Beutler testi ile GALT aktivitesi değerlendirilebilir. Laboratuvar bulguları arasında yüksek transaminaz, hiperbilirubinemi, hipoalbüminemi, koagülopati, idrarda galaktoz ve aminoasidüri bulunur. Kesin tanı eritrositlerde GALT aktivitesinin ölçülmesi, Gal-1-P düzeylerinin saptanması ve genetik analiz ile konur.
10) Ayırıcı Tanılar
Konjenital enfeksiyonlar, neonatal hemokromatoz, herediter tirozinemi, mitokondriyal hastalıklar ve Fanconi-Bickel sendromu ayırıcı tanıda göz önünde bulundurulmalıdır.
11) Tedavi
Şüphe halinde dahi galaktoz ve laktoz kısıtlı diyete geçilmelidir. Tedavi ömür boyu devam eder. Soya bazlı formüller önerilir. Süt ve süt ürünleri tamamen yasaktır. Laktozsuz süt ürünleri galaktoz içerdiğinden uygun değildir. Bazı sert peynirler eser miktarda galaktoz içerdiğinden kontrollü tüketilebilir. Kalsiyum ve D vitamini takviyesi gereklidir.
12) Prognoz
Erken tanı ve tedavi ile akut mortalite azalır. Ancak endojen galaktoz üretimi nedeniyle diyet tedavisine rağmen uzun dönem komplikasyonlar tamamen önlenemez. Nörogelişimsel sorunlar ve kadınlarda gonadal yetmezlik en önemli uzun dönem sekel gruplarını oluşturur.
13) Güncel Araştırmalar
Yeni tedavi yaklaşımları arasında aldoz redüktaz inhibitörleri (ör. govorestat) dikkat çekmektedir. Bu ilaçlar galaktitol birikimini azaltmayı hedeflemektedir. Klinik araştırmalar umut verici sonuçlar bildirse de henüz onaylanmış spesifik bir farmakolojik tedavi bulunmamaktadır.
Keşif
İlk Klinik Gözlemler (1900–1935)
- yüzyılın başlarında bazı bebeklerin süt içtikten sonra hızla sarılık, hepatomegali, letarji ve ölümcül seyirli sepsis geliştirdiği fark edildi. Bu durum başlangıçta “idiyopatik neonatal hepatit” benzeri tablolarla karıştırıldı. Ancak klinisyenler giderek bu olguların ortak yönünün süt veya süt ürünleriyle ilişkili olduğunu gözlemlemeye başladılar.
Goppert’in Tanımı (1935)
Galaktozemi, bağımsız bir klinik varlık olarak ilk kez 1935’te Goppert tarafından tanımlandı. O, galaktoz içeren besinlerle temas sonrası ağır toksik reaksiyonlar gösteren bebekleri sistematik biçimde rapor etti. Böylece hastalık literatüre “Galaktosämie” adıyla girdi.
Biyokimyasal Keşifler (1940–1950)
İkinci Dünya Savaşı sonrasında metabolik bozukluklara yönelik biyokimyasal araştırmalar ivme kazandı. 1940’ların sonlarından itibaren araştırmacılar, galaktozun organizmada glikoza çevrildiğini ve bu dönüşümde özel enzimlerin rol aldığını ortaya koydular.
Leloir ve Leloir Yolu (1956)
1956’da Luis Federico Leloir ve ekibi, galaktoz metabolizmasının üç basamaklı enzimatik yolunu tanımladı. Bu yol daha sonra “Leloir Yolu” olarak anıldı. Çalışmaları galaktoz metabolizmasının temel mekanizmasını açıklığa kavuşturdu ve kendisine 1970 Nobel Kimya Ödülü’nü kazandırdı.
Enzimatik Kusurların Ayırt Edilmesi (1960–1970)
1960’larda araştırmacılar farklı galaktozemi tiplerini tanımlamaya başladılar. Galaktoz-1-fosfat üridil transferaz (GALT) eksikliğinin klasik galaktozemiden sorumlu olduğu gösterildi. Daha sonra galaktokinaz (GALK) eksikliği ile infantil katarakt olguları ilişkilendirildi. UDP-galaktoz 4-epimeraz (GALE) eksikliğinin ise daha nadir ama değişken klinik tablolara yol açtığı ortaya kondu.
Tanı Yöntemlerinin Gelişmesi (1970–1980)
Bu dönemde eritrositlerde enzim aktivitesini ölçen laboratuvar testleri standart hale geldi. İdrarda indirgeme testleri ve Gal-1-P düzeylerinin ölçülmesi tanıda kritik hale geldi. Böylece galaktozemi, diğer neonatal karaciğer yetmezliği sendromlarından ayırt edilebilir hale geldi.
Moleküler Dönüşüm (1980–1990)
1980’lerin sonundan itibaren moleküler biyoloji tekniklerinin gelişmesiyle GALT geninin klonlanması ve mutasyon analizleri mümkün oldu. İlk kez spesifik mutasyonların (örneğin p.Q188R, p.K285N) klasik galaktozemi ile ilişkili olduğu gösterildi. Aynı dönemde Duarte varyantı gibi hafif formlar da tanımlandı.
Yenidoğan Tarama Programları (1990’lar)
1990’lardan itibaren birçok ülkede yenidoğan tarama panellerine galaktozemi dahil edilmeye başlandı. Bu sayede hastalık, semptomlar gelişmeden tanınabilir hale geldi ve akut mortalite önemli ölçüde azaldı. Ancak uzun dönem komplikasyonların tarama ve erken tedaviye rağmen devam etmesi, hastalığın biyolojisinin daha karmaşık olduğunu gösterdi.
2000’lerden Günümüze
2000’li yıllarda uluslararası hasta kayıtları ve araştırma ağları kuruldu. Tanı algoritmaları daha sofistike hale geldi; moleküler testler rutinleşti. Farmakolojik tedavi araştırmaları (örneğin aldoz redüktaz inhibitörleri) gündeme geldi. 2020’li yıllarda klinik araştırmalar, galaktitol birikimini azaltmaya yönelik tedavilerle yeni bir umut vadetmeye başladı.
İleri Okuma
- Goppert F. (1935). Über hereditäre Galaktosämie. Klinische Wochenschrift, 14: 473–475.
- Kalckar HM. (1957). Galactose metabolism and cell physiology: a retrospective. Federation Proc., 16(3): 818–825.
- Leloir LF. (1956). The enzymatic transformation of galactose into glucose derivatives. Arch Biochem Biophys, 65(2): 378–390.
- Beutler E, Baluda MC. (1966). A simple spot screening test for galactosemia. J Lab Clin Med, 68(1): 137–141.
- Elsas LJ, Dembure PP. (1985). Molecular genetics of galactosemia. Hum Genet, 70(1): 1–5.
- Elsas LJ, Lai K. (1998). The molecular biology of galactosemia. Genet Med, 1(1): 40–48.
- Bosch AM. (2006). Classical galactosaemia revisited. J Inherit Metab Dis, 29(4): 516–525.
- Welling L, et al. (2017). International clinical guideline for the management of classical galactosemia. J Inherit Metab Dis, 40(2): 171–176.
- Rubio-Gozalbo ME, et al. (2019). The natural history of classic galactosemia: lessons from the GalNet registry. J Inherit Metab Dis, 42(5): 718–727.
- Berry GT, Fridovich-Keil JL. (2023). Classic Galactosemia and Clinical Variant Galactosemia (GALT). GeneReviews®.
- Fridovich-Keil JL, Bean LJH, He M, Schroer R. (2011/2021 günc.) Epimerase Deficiency Galactosemia (GALE). GeneReviews®.
- Grünert SC, et al. (2012). Fanconi–Bickel syndrome: GLUT2 mutations associated with a distinct clinical phenotype. Mol Genet Metab, 105(3):368–372.
- Kehar M, et al. (2014). Fanconi–Bickel Syndrome – Mutation in SLC2A2 Gene. Case Rep, PMC4353876.
- Welling L, Bernstein LE, Berry GT, et al. (2017). International clinical guideline for the management of classical galactosemia: diagnosis, treatment and follow-up. J Inherit Metab Dis, 40(2):171–176.
- Demirbas D, et al. (2018). Hereditary galactosemia. Metabolism, 83:188–196.
- Rubio-Gozalbo ME, et al. (2019). The natural history of classic galactosemia. J Inherit Metab Dis, PMC6486996.
- Delnoy B, et al. (2021). Current and future treatments for classic galactosemia. Genes, PMC7911353.
- Badiu Tișa I, et al. (2022). The Importance of Neonatal Screening for Galactosemia. Nutrients, 15(1):10.
- Berry GT, Fridovich-Keil JL. (2023). Classic Galactosemia and Clinical Variant Galactosemia (GALT). GeneReviews®.
- Walter JH, Fridovich-Keil JL. (2024). Galactosemia. In: The Online Metabolic and Molecular Bases of Inherited Disease (OMMBID).
- Bailey E, et al. (2025). Results of the ACTION-Galactosemia Kids Study to Evaluate Govorestat. J Clin Pharmacol.