
Hipertirozinemi, kan plazmasında yüksek düzeyde amino asit tirozin ile karakterize bir grup metabolik bozukluğu ifade eder. Bu durum, tirozin metabolizmasından sorumlu enzimlerdeki genetik kusurlardan kaynaklanır ve vücutta birikmesine neden olur. Farklı hipertirozinemi türleri, etkilenen spesifik enzime göre sınıflandırılır ve her biri farklı klinik ve patofizyolojik özellikler gösterir.

Hipertirozinemi Sınıflandırması
Hipertirozinemiler, tirozin metabolizması yolundaki hangi enzimin eksik olduğuna bağlı olarak üç ana tipte sınıflandırılır:
Tip I Hipertirozinemi (Tirozinemi Tip I)
- Enzim defekti**: Tirozin katabolik yolundaki son enzim olan *fumarylacetoacetate hydrolase (FAH)* enziminin eksikliğinden kaynaklanır.
- Patofizyoloji**: Defekt, *fumarylacetoacetate* ve succinylacetone gibi toksik metabolitlerin birikmesine yol açarak ciddi karaciğer ve böbrek hasarına neden olur.
- Klinik tablo**: Belirtiler genellikle bebeklik döneminde ortaya çıkar ve *gelişememe, sarılık, hepatomegali (karaciğer büyümesi), kusma* ve nörolojik krizleri içerebilir. Tedavi edilmezse karaciğer yetmezliğine, renal tübüler disfonksiyona ve hatta hepatoselüler karsinoma yol açabilir.
Tip II Hipertirozinemi (Tirozinemi Tip II)
- Enzim defekti**: Tirozini *p-hidroksifenilpiruvata* dönüştüren tirozin aminotransferaz (TAT) eksikliğinden kaynaklanır.
- Patofizyoloji**: Defekt, yüksek tirozin seviyelerine ve dokularda birikime yol açar, özellikle cildi, gözleri ve sinir sistemini etkiler.
- Klinik sunum**: Hastalar genellikle avuç içlerinde ve ayak tabanlarında *ağrılı deri lezyonları*, *fotofobi (ışık hassasiyeti)* ve keratit gibi göz sorunları ve bazı durumlarda zihinsel engellilik sergiler. Belirtiler tipik olarak erken çocukluk döneminde ortaya çıkar.
Tip III Hipertirozinemi (Tirozinemi Tip III)
- Enzim defekti**: Bu form, *p-hidroksifenilpiruvatın* homogentisata dönüşümünü katalize eden 4-hidroksifenilpiruvat dioksijenaz (HPD) eksikliğinden kaynaklanır.
- Patofizyoloji**: Bu durum *p-hidroksifenilpiruvat* ve tirozin birikimine yol açarak nörolojik ve gelişimsel sorunlara neden olabilir.
- Klinik sunum**: Belirtiler Tip I ve II’ye kıyasla daha hafiftir ve *zihinsel engeller, nöbetler* ve aralıklı ataksi içerebilir.

Patofizyoloji ve Semptomlar
Hipertirozineminin tüm formları, 1,25 mg/dL’yi aşan ve idrarda tirozin atılımına (tirozinüri) yol açan yüksek plazma tirozin seviyeleri ortak özelliğini paylaşır. Tirozin ve metabolitlerinin birikimi çeşitli sistemik etkilere neden olabilir:
- Karaciğer tutulumu**: Tip I’de, *fumarilasetoasetat* gibi toksik metabolitlerin birikimi önemli karaciğer hasarına neden olarak hepatomegali, siroz, karaciğer yetmezliği ve hatta hepatoselüler karsinoma yol açar. Sarılık ve koagülopati de görülebilir.
- Böbrek fonksiyon bozukluğu**: Tip I’de toksik metabolitler *böbrek tübüllerine* zarar vererek böbrek tübüler asidozuna neden olabilir ve böbreğin belirli maddeleri atma yeteneğini bozabilir.
- Nörolojik semptomlar**: Tip I’deki nörolojik krizler *ağrı, halsizlik, nöbetler* ve kendini yaralama davranışları içerebilir. Tip II ve III’te bilişsel bozukluk, nöbetler veya hareket bozuklukları görülebilir.
- Cilt ve göz semptomları**: Tip II’de, *el ve ayak tabanlarında* ağrılı hiperkeratotik plakların** yanı sıra keratit** ve fotofobi gibi göz tahrişleri yaygın olarak görülür.
- İskelet tutulumu**: Tirozin metabolizması bozuklukları, özellikle Tip I’de, bozulmuş D vitamini metabolizması ve renal tübüler disfonksiyon nedeniyle *riketlere* yol açabilir.
- Melanin metabolizması bozuklukları**: Tirozin, melanin üretiminde bir öncüdür. Yüksek tirozin seviyeleri normal *melanin sentezini* bozarak pigmentasyon sorunlarına yol açabilir.

Teşhis
Hipertirozinemi teşhisi şunları içerir:
- Kan testleri**: Yüksek *plazma tirozin seviyeleri* önemli bir belirteç görevi görür.
- İdrar organik asit analizi**: Tip I’de *süksinilaseton* varlığı tanı koydurucudur.
- Genetik test: Eksik enzimlerden sorumlu genlerdeki FAH, TAT veya HPD gibi mutasyonların tanımlanması.
- Karaciğer ve böbrek fonksiyon testleri**: Özellikle Tip I’de organ tutulumunun derecesinin değerlendirilmesi.
- Görüntüleme çalışmaları**: Karaciğer ve böbrek hasarını değerlendirmek için *Ultrason* veya MRI.
Terapi ve Yönetim
Enzim kusurlarını tamamen düzeltebilecek nedensel bir tedavi bulunmamakla birlikte, çeşitli terapötik stratejiler semptomları yönetmeye ve komplikasyonları azaltmaya yardımcı olabilir:
Diyet Yönetimi
- Tedavinin temel taşı düşük proteinli diyet, özellikle fenilalanin ve tirozin bakımından zengin gıdaların (örn. süt ürünleri, et, balık, soya, fındık) kısıtlanmasıdır. Bu, kan tirozin seviyelerini düşürmeye ve toksik metabolitlerin birikimini azaltmaya yardımcı olur.
- Metabolik bozukluklar için özel olarak tasarlanmış Tıbbi gıdalar veya formüller aşırı tirozin veya fenilalanin olmadan gerekli besinleri sağlamak için kullanılabilir.
Farmakolojik Tedavi
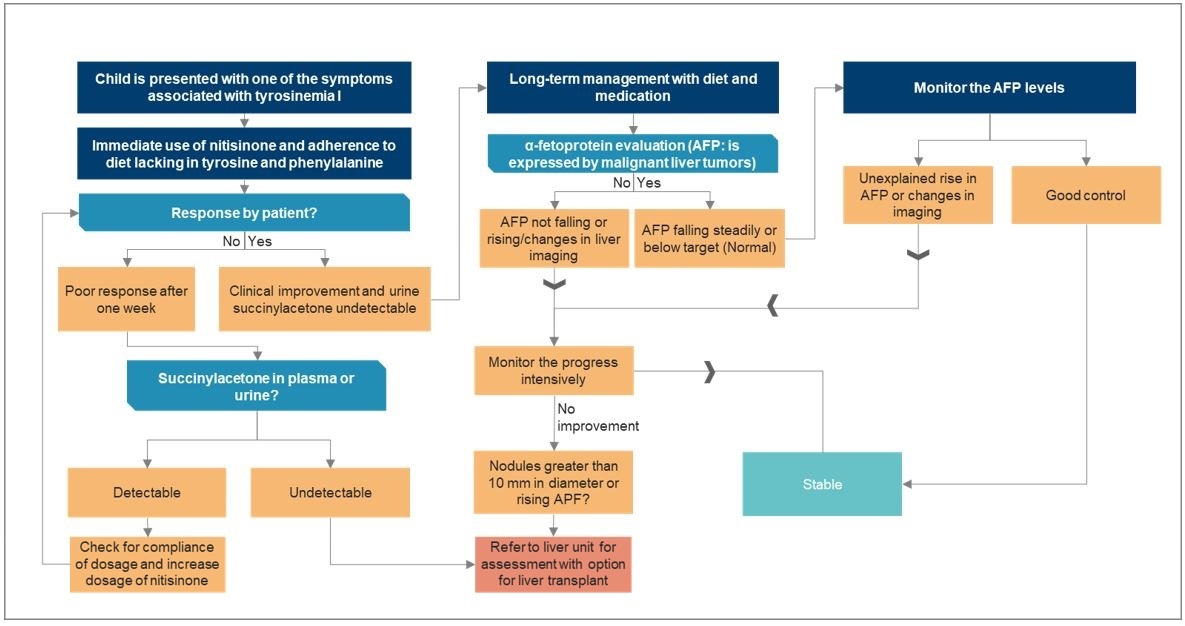
- Nitisinon (NTBC)** Tirozinemi Tip I için birincil tedavidir. 4-hidroksifenilpiruvat dioksijenazı** inhibe ederek süksinilaseton gibi toksik metabolitlerin oluşumunu engeller. Bu ilaç karaciğer ve böbrek fonksiyonlarını önemli ölçüde iyileştirebilir ve karaciğer kanseri riskini azaltabilir.
- Böbrek fonksiyon bozukluğuyla ilişkili raşitizm sorununu çözmek için D vitamini takviyesi gerekli olabilir.
Karaciğer Nakli
- Ağır Tirozinemi Tip I vakalarında, özellikle nitisinon tedavisi başarısız olduğunda veya karaciğer kanseri geliştiğinde, karaciğer nakli düşünülebilir. Metabolik bozukluğu düzelten hayat kurtarıcı bir tedavi olabilir.
İzleme ve Destekleyici Bakım
- Karaciğer ve böbrek fonksiyonlarının**, *nörolojik durumun* ve büyüme parametrelerinin düzenli olarak izlenmesi esastır.
- Tip II** hastalarında göz semptomlarını erken tespit etmek ve yönetmek için göz muayeneleri önemlidir.
- Bilişsel veya öğrenme güçlüğü olan çocuklar için gelişimsel değerlendirmeler ve eğitim desteği gerekebilir.
İçindekiler
Prognoz
Hipertirozineminin prognozu büyük ölçüde tipine ve tedavinin zamanına bağlıdır:
- Tip I Hipertirozinemi** erken nitisinon tedavisi ve diyet yönetimi ile daha iyi bir prognoza sahiptir, ancak karaciğer kanseri riski bir endişe kaynağı olmaya devam etmektedir.
- Tip II ve III** genellikle daha az şiddetlidir ve diyet kısıtlamaları ile yönetilebilir, ancak göz bakımı ve gelişimsel destek gerekebilir.
Keşif
Hipertirozinemi araştırmalarının hikayesi, tıbbi zorluklar, bilimsel atılımlar ve hayat kurtaran dönüştürücü tedavilerden biridir. Tarih boyunca, önemli dönüm noktaları bu karmaşık metabolik bozukluk grubunun anlaşılması ve yönetilmesinde kaydedilen ilerlemeyi işaret etmektedir.
İlk Günler: Gizemli Bir Durumun Tanınması (1930’lar-1950’ler)
Tirozin metabolizmasıyla ilgili bir durumun ilk belirtileri 1930’larda doktorların bazı bebeklerin tuhaf semptomlar gösterdiğini fark etmesiyle ortaya çıkmaya başladı: Gelişememe, kusma, sarılık ve raşitizm benzeri kemik deformiteleri. Bu bebeklerde ayrıca doktorları şaşırtan ve altta yatan metabolik bir bozukluğa işaret eden lahana benzeri belirgin bir koku vardı. Ancak, bu semptomların kesin nedeni anlaşılamamıştır.
1950’lere** gelindiğinde, biyokimyasal analizdeki gelişmeler araştırmacıların etkilenen hastaların kan ve idrarında yüksek seviyelerde tirozin tespit etmelerine olanak sağladı. Bu, hastalığı amino asit metabolizmasındaki bir bozukluğa bağlayan önemli bir ipucuydu.** Yine de, hangi enzimlerin dahil olduğunu ve bu metabolik bozulmanın nasıl meydana geldiğini anlamak hala bir gizemdi. Bilgi eksikliği, doktorlara tedavi için sınırlı seçenekler bırakıyor, öncelikle semptomatik yönetim ve diyet ayarlamalarına odaklanıyordu.
Bir Dönüm Noktası: Tirozinemi Tip I’in Tanımlanması (1960’lar-1970’ler)
Araştırmacıların Tirozinemi Tip I hastalığını ayrı bir metabolik bozukluk olarak tanımlayabildikleri 1960’larda** bu alanda önemli bir adım atılmıştır. Bu ilerleme büyük ölçüde, ciddi karaciğer ve böbrek sorunları yaşayan çocuklarda vakaları belgeleyen ve sonuçta durumu **tirozin ve toksik yan ürünlerinin birikimine bağlayan *Dr. Reye ve ekibinin* çabaları sayesinde gerçekleşti.
Tirozin yıkımının son aşamasında yer alan bir enzim olan fumarilasetoasetat hidrolazın (FAH) bu hastalarda eksik veya kusurlu olduğunun anlaşılması 1970’lerde çığır açan bir keşif oldu. Bu eksiklik, süksinilaseton gibi toksik metabolitlerin neden vücutta birikerek hayati organlara zarar verdiğini açıkladı. FAH eksikliğinin Tirozinemi Tip I’in nedeni olarak tanımlanması yalnızca tanısal doğruluğu artırmakla kalmadı, aynı zamanda altta yatan metabolik yolların anlaşılmasına da kapı açarak gelecekteki tedavilerin önünü açtı.
Tip II ve Tip III Tanımlandı: Sınıflandırmanın Genişletilmesi (1970’ler-1980’ler)
Araştırmacılar tirozin metabolizmasını daha derinlemesine inceledikçe, farklı enzim kusurlarının farklı hipertirozinemi türlerine yol açabileceği anlaşıldı. 1970’lerde** klinisyenler, ellerde ve ayaklarda ağrılı cilt lezyonları, ışık hassasiyeti ve göz problemleri ile karakterize, hastalığın daha hafif bir formu olan Tirozinemi Tip II’yi tanımaya başladılar.** Bundan sorumlu enzim kusurunun, tirozin yıkımında daha erken bir adımı katalize eden tirozin aminotransferazda (TAT) olduğu bulundu.
Tirozinemi Tip II’nin** hikayesi, yıllardır ağrılı keratit ve cilt sorunlarından muzdarip olan kırsal bir çiftçi topluluğundan genç bir çocuğun dahil olduğu bir vaka ile dokunaklı bir hal aldı. Semptomları kandaki yüksek tirozin seviyeleriyle ilişkilendirilene kadar doğru teşhis konulamamış, bu da aileye rahatlama ve düşük proteinli bir diyetle hedefe yönelik bir yönetim stratejisi sunmuştur.
1980’lerde,** araştırmacılar **4-hidroksifenilpiruvat dioksijenaz (HPD) eksikliğine bağlı daha nadir ve daha hafif bir form olan *Tirozinemi Tip III’ü* tanımladılar.** Bu durum genellikle nöbetler ve aralıklı ataksi gibi nörolojik semptomlar olarak ortaya çıktı ve tirozin yıkım yolu boyunca çeşitli enzim eksikliklerinin farklı klinik tablolara neden olabileceği sonucuna yol açtı.
Oyunu Değiştiren: Nitisinon Bir Tedavi Olarak Ortaya Çıkıyor (1992)
Araştırmacılar, tarımda kullanılan ve nitisinon (NTBC) olarak bilinen bir herbisitin **4-hidroksifenilpiruvat dioksijenaz enzimini inhibe edebildiğini keşfettiğinde *1992* yılında dönüştürücü bir atılım gerçekleşti. ** İlginç bir şekilde, bu enzim tirozin yıkımının erken bir adımında yer alır ve Tirozinemi Tip I’deki metabolik bloğun akış yukarısında yer alır. Nitisinon bu enzimi inhibe ederek, süksinilaseton gibi toksik metabolitlerin** oluşumunu etkili bir şekilde **önledi, böylece karaciğer ve böbrekleri hasardan korudu.
Keşif tesadüfi oldu: nitisinonun herbisit özelliklerini inceleyen bilim insanları, bitkilerde tirozin metabolizmasını bozduğunu fark etti. Denemeler nitisinonun plazma süksinilaseton seviyelerini önemli ölçüde düşürdüğünü ve etkilenen hastalarda karaciğer fonksiyonlarını iyileştirdiğini gösterdiğinde, hastalığın tedavisinde devrim yarattı. Pek çok hasta için bir zamanlar ölümcül bir tanı olan bu durum artık çok daha iyi bir prognoza sahip yönetilebilir bir kronik durum haline gelmişti.
Genetik Bilgiler ve Prenatal Tanı: 21. Yüzyıl
2000’li yılların başları**, *DNA dizileme teknolojilerindeki* ilerlemelerin üç tip hipertirozinemiden sorumlu FAH, TAT ve HPD genlerindeki mutasyonların tanımlanmasını sağlamasıyla genetik anlayışta yeni bir çağ başlattı. Genetik testler daha doğru tanıya, taşıyıcı taramasına ve hatta doğum öncesi tanıya olanak tanıyarak ailelere planlama ve erken yönetim için önemli bilgiler sağladı.
Bu dönemden özellikle dokunaklı bir hikaye, ilk çocuklarını teşhis edilmemiş Tirozinemi Tip I nedeniyle kaybettikten sonra, ikinci çocuklarını hamileliğin erken dönemlerinde teşhis etmek için prenatal genetik test kullanan bir çifti içeriyordu. Doğumdan sonra derhal nitisinon tedavisine başlanması sayesinde bebek sağlıklı bir şekilde büyümüş ve ilk çocuklarının hayatını kaybetmesine neden olan ciddi komplikasyonlardan kurtulmuştur. Bu hikaye, modern genetik tıbbın metabolik bozukluklardan etkilenen aileler üzerindeki derin etkisini yansıtmaktadır.
Gelecek: İyileştirici Tedavilere Doğru
Günümüzde araştırmacılar, hipertirozinemi için potansiyel iyileştirici yaklaşımlar olarak gen tedavisi ve enzim replasman tedavisini araştırmaktadır. Deneysel çalışmalar, altta yatan genetik kusurları düzeltmeyi amaçlamakta ve hastaların ömür boyu diyet kısıtlamalarına veya farmakolojik tedavilere güvenmek zorunda kalmayabilecekleri bir gelecek için umut vermektedir.
Hipertirozineminin öyküsü, gizemli bir metabolik durumdan etkili tedavilerin hasta sonuçlarını dönüştürdüğü bir duruma evrilen dikkate değer bir ilerlemedir. Tıpta bilimsel keşif, tesadüf ve inovasyonun gücünün bir kanıtı olarak, bu nadir ama ciddi metabolik bozukluklardan etkilenenler için daha parlak bir gelecek sunuyor.
İleri Okuma
- Lindstedt, S., Holme, E., Lock, E. A., Hjalmarson, O., & Strandvik, B. (1992). “Treatment of hereditary tyrosinaemia type I by inhibition of 4-hydroxyphenylpyruvate dioxygenase.” The Lancet, 340(8823), 813-817.
- Mitchell, G., Grompe, M., Lambert, M., & Tanguay, R. M. (2001). “Hypertyrosinemia.” The Metabolic and Molecular Bases of Inherited Disease, 1777-1805.
- Russo, P. A., Mitchell, G. A., & Tanguay, R. M. (2001). “Tyrosinemia: a review.” Pediatric Pathology & Molecular Medicine, 20(4), 249-272.
- Scott, C. R. (2006). “The genetic tyrosinemias.” American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 142C(2), 121-126.
- de Laet, C., Dionisi-Vici, C., Leonard, J. V., McKiernan, P., Mitchell, G., & Saudubray, J. M. (2013). “Recommendations for the management of tyrosinaemia type 1.” Orphanet Journal of Rare Diseases, 8, 8.