İçindekiler
ICD-10 Sınıflandırması
- C90.1 – Plazma hücre lösemisi
- C91 – Lenfoid (lenfoblastik/lenfositik) lösemiler
- C92 – Miyeloid lösemiler
- C93 – Monositik lösemiler
- C94 – Diğer belirtilmiş lösemiler
- C95 – Belirlenmemiş hücre tipli lösemi
Tanım ve Temel Sınıflandırma
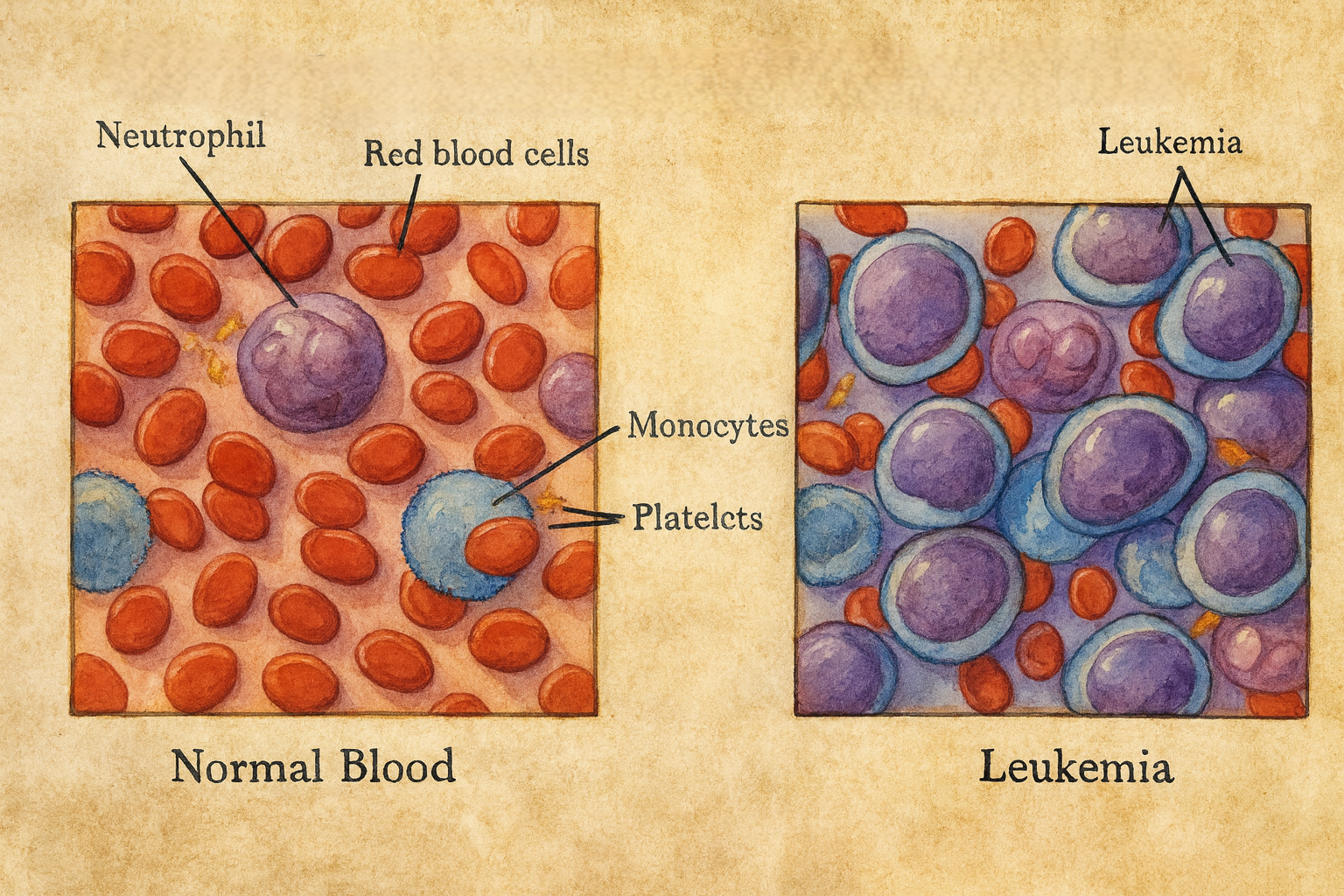
Lösemi, kemik iliğinde yer alan hematopoetik kök/öncül hücrelerin klonal malign proliferasyonu ile karakterize bir grup hastalığın üst başlığıdır. Patolojik klon, normal hematopoezi bastırarak anemi, trombositopeni ve nötropeniye; dolaşıma yayılarak periferik kanda blast artışına ve çeşitli organlarda infiltrasyona yol açar. Hastalığın etimolojik kökeni “leukos (beyaz) + haima (kan)” birleşiminden gelir; bu, özellikle akut tabloda periferik beyaz küre artışıyla tarihsel olarak gözlenen “beyaz kan” görünümüne atıftır.
Dört ana klinik-biyolojik tip:
- Akut lenfoblastik lösemi (ALL)
- Akut miyeloid lösemi (AML)
- Kronik lenfositik lösemi (KLL)
- Kronik miyeloid lösemi (KML)
“Akut” tablolar blastik hücrelerin hızlı proliferasyonu ve >%20 kemik iliği blast oranıyla (bazı istisnai translokasyonlarda daha düşük eşikle) seyrederken, “kronik” tablolar daha matür hücre birikimi ve görece yavaş ilerleyişle karakterizedir.
Epidemiyoloji ve Risk Etmenleri
Lösemiler tüm yaş gruplarında görülebilir; ALL çocukluk çağının en sık malignitesidir, AML erişkinlerde en sık akut lösemidir. KLL ileri yaşta sık görülür; KML her yaşta olabilir, sıklıkla orta yaş-ileri yaşta saptanır.
Çevresel ve edinsel etmenler
- İyonlaştırıcı radyasyon (yüksek doz, terapötik/mesleki maruziyet)
- Kimyasal ajanlar (özellikle benzen ve bazı organik çözücüler)
- Kemoterapötikler (alkilleyiciler; topoizomeraz II inhibitörleri ile tedavi ilişkili AML)
- Enfeksiyöz etmenler (özellikle HTLV-1 erişkin T-hücreli lösemi/lenfoması ile)
- Sigara ve bazı mesleki maruziyetler (tarım, boya, petrokimya)
Genetik duyarlılık ve predispozisyon sendromları
- Down sendromu (özellikle megakaryoblastik AML), Fanconi anemisi, Bloom sendromu, Ataksi-telenjiektazi
- Germline yatkınlıklar: RUNX1, CEBPA, GATA2, DDX41 mutasyonları
- Yaşla ilişkili klonal hematopoez (CHIP): DNMT3A, TET2, ASXL1 gibi sürücü mutasyonların subklinik klonları ileride MDS/AML riskini artırabilir.
Moleküler Patogenez
Lösemi gelişiminde genetik ve epigenetik olayların kademeli birikimi belirleyicidir:
- Kromozomal yeniden düzenlenmeler (translokasyonlar):
- t(9;22)(q34;q11) – BCR-ABL1 (Philadelphia): KML’nin ayırt edici lezyonu; ayrıca Ph-pozitif ALL.
- t(15;17)(q24;q21) – PML-RARA: Akut promiyelositik löseminin (APL) temel lezyonu.
- t(8;21), inv(16)/t(16;16), 11q23/KMT2A yeniden düzenlenmeleri (AML alt tipleri).
- Nükleotid metabolizması ve epigenetik düzenleyiciler:
IDH1/IDH2 mutasyonları (AML’de bir alt grupta), NPM1, FLT3-ITD/TKD, DNMT3A, TET2, ASXL1 vb.
IDH mutasyonları 2-hidroksiglutarat birikimiyle epigenetik programı bozarak farklılaşmayı engeller. - Sinyal iletimi:
BCR-ABL1 tirozin kinaz aktivitesi; JAK-STAT, MAPK, PI3K/AKT yollarında kazanılmış aktivasyon.
Bu lezyonlar proliferasyon, apoptozdan kaçış, öz-yenilenme artışı ve diferansiyasyon blokajı ile sonuçlanır; ortaya çıkan klon, kemik iliğinde nişi işgal ederek normal hematopoezi baskılar.
Klinik Tablo
Belirti ve bulgular, kemik iliği yetmezliği ve doku infiltrasyonu ekseninde toplanır. Klinik heterojenlik geniştir; bazı kronik formlar uzun süre belirti vermeyebilir.
Semptomlar ve bulgular
- Anemi: Halsizlik, solukluk, efor dispnesi, taşikardi
- Trombositopeni: Peteşi, purpura, kolay morarma, mukozal kanamalar
- Nötropeni: Ateş, tekrarlayan/opportunistik enfeksiyonlar
- Kemiğe bağlı: Kemik ağrısı, hassasiyet; özellikle çocuk ALL’de
- Organomegali: Hepatomegali-splenomegali (sık), lenfadenopati (özellikle ALL/KLL’de)
- Mediastinal kitle/timus büyümesi: T-hücre ALL’de görülebilir
- Ekstramedüller tutulum: Deri (leukemia cutis), dişeti hipertrofisi (monositik AML), SSS/CNS ve testis tutulumları (ALL’de)
- Leukostaz (yüksek lökosit yükü): Baş ağrısı, görme bozukluğu, dispne, nörolojik semptomlar
- Tümör lizis sendromu: Spontan veya tedavi sonrası; hiperürisemi, hiperkalemi, hiperfosfatemi, hipokalsemi
Not: “Lösemid reaksiyon” (ağır enfeksiyon/iltihapta reaktif lökositoz ve genç hücre artışı) gerçek lösemiden morfoloji, akım sitometrisi ve sitogenetik ile ayırt edilmelidir.
Tanı Yaklaşımı
- Tam kan sayımı ve periferik yayma: Anemi, trombositopeni, lökositoz ya da lökopeni; blastların varlığı.
- Kemik iliği aspirasyon/biopsisi: Hücresellik, blast oranı; akut lösemide genellikle blast ≥%20.
- Akım sitometrisi (immünfenotipleme): B-ALL (CD19, CD22, CD10), T-ALL (CD3, CD7), AML (CD13, CD33, MPO), KLL (CD19, CD5, CD23 ko-ekspresyonu) gibi imzalar.
- Sitogenetik ve moleküler testler: Karyotip, FISH; PCR/NGS ile sürücü mutasyonlar (BCR-ABL1, NPM1, FLT3, IDH1/2 vb.).
- Biyokimya: LDH, ürik asit, elektrolitler; tümör lizis ve organ fonksiyonları.
- Görüntüleme: Mediastinal genişleme (T-ALL), organomegali; klinik gerekçeyle.
- SSS değerlendirmesi: Özellikle ALL’de lomber ponksiyon ile BOS sitolojisi.
Risk sınıflaması (alt tipe özgü):
- AML için ELN risk grupları (sitogenetik/moleküler temelli),
- KML için Sokal/Hasford/EUTOS skorları + BCR-ABL1 moleküler yanıt düzeyleri,
- KLL için IGHV mutasyon durumu, TP53/17p delesyonu, kompleks karyotip,
- ALL için yaş, lökosit sayısı, sitogenetik (Ph+, KMT2A vb.), asgarî rezidüel hastalık (MRD).
Alt Tiplere Özgü Özellikler
Akut Lenfoblastik Lösemi (ALL)
- Epidemiyoloji: Çocukluk çağında pik; erişkinde prognoz görece daha kötü.
- Klinik: Ateş, kanama, kemik ağrısı; lenfadenopati ve hepatosplenomegali sık. T-ALL’de mediastinal kitle ve superior vena kava sendromu olabilir.
- Biyoloji: B-hücre ve T-hücre soyları; Ph-pozitif ALL erişkinde anlamlı bir alt gruptur.
- Tedavi:
- Çok aşamalı kombinasyon kemoterapisi (indüksiyon-konsolidasyon-idame).
- MRD kılavuzlu tedavi uyarlamaları.
- Ph-pozitif ALL: Kemoterapiye tirozin kinaz inhibitörü (TKİ) eklenir.
- Nüks/dirençli hastalık: Blinatumomab (CD19-BiTE), Inotuzumab ozogamicin (CD22-konjugat), CD19 CAR-T seçenekleri; uygun hastaya allojenik kök hücre nakli.
Akut Miyeloid Lösemi (AML)
- Epidemiyoloji: Erişkinlerde en sık akut lösemi; yaşla insidans artar.
- Klinik/Biyoloji: Monositik alt tipte dişeti hipertrofisi ve cilt tutulumu; moleküler heterojenite belirgindir (NPM1, FLT3, IDH1/2, RUNX1 vb.).
- Tedavi:
- Standart indüksiyon: “7+3” (sitarabin + antrasiklin) veya hastaya özgü modifikasyonlar.
- Hedefe yönelik: FLT3 inhibitörleri, IDH1/2 inhibitörleri, ikili kombinasyonlar; sekonder AML’de CPX-351.
- Yaşlı/komorbid hastalarda: Hipometilleyici ajan + venetoklaks rejimleri.
- Uygun adayda: Allojenik nakil; yüksek risk ya da MRD-pozitiflikte özellikle.
Akut Promiyelositik Lösemi (APL, AML-M3)
- Genetik: t(15;17) / PML-RARA.
- Özellik: DİK eğilimi ile acil tanı/tedavi gerektirir.
- Tedavi: ATRA + arsenik trioksit (kemoterapisiz yaklaşım olası); koagülopati yönetimi kritik.
Kronik Miyeloid Lösemi (KML)
- Genetik imza: BCR-ABL1 pozitifliği (Philadelphia kromozomu).
- Fazlar: Kronik faz → hızlanmış faz → blastik kriz (akut lösemi benzeri).
- Tedavi:
- TKİ’ler (ör. imatinib, dasatinib, nilotinib, bosutinib, ponatinib).
- Derin ve kalıcı moleküler yanıtla bazı olgularda tedavisiz tedavi başarısı (TFR) hedeflenebilir.
- Direnç/yan etkide ikinci/üçüncü kuşak TKİ seçenekleri; nadiren nakil.
Kronik Lenfositik Lösemi (KLL)
- Epidemiyoloji: İleri yaşta en sık kronik lösemi; çoğu zaman tesadüfen saptanır.
- Biyoloji: IGHV mutasyon durumu, TP53/17p değişiklikleri prognostik önemdedir.
- Yaklaşım:
- Belirtisiz hastada izlem (“watch-and-wait”).
- Tedavi gerektirenlerde:
- BTK inhibitörleri (ibrutinib, akalabrutinib, zanubrutinib),
- BCL-2 inhibitörü venetoklaks + anti-CD20 (obinutuzumab/rituksimab) sabit süreli rejimler,
- Uygun hastada kombinasyon/ardışık stratejiler; nadiren nakil.
Plazma Hücre Lösemisi (C90.1)
- Tanım: Dolaşımda belirgin plazma hücresi varlığı ile seyreden, çoğu zaman multipl miyelom spektrumunda yer alan agresif tablo.
- Tedavi: Proteazom inhibitörleri, immünomodülatörler, anti-CD38 antikorları, uygun hastada yüksek doz tedavi + otolog/allo nakil; sıkı enfeksiyon profilaksisi.
Komplikasyonlar
- Enfeksiyonlar: Nötropeniye bağlı; bakteriyel, fungal (invaziv aspergilloz, kandidiyaz), viral reaktivasyonlar.
- Kanama: Trombositopeni ve APL’de DİK.
- Leukostaz: Hiperviskozite, solunum/nörolojik bulgular; acil lökaferez ve sitoredüksiyon gerekebilir.
- Tümör lizis sendromu: Böbrek yetmezliği riski; profilaksi ve hızlı destek şarttır.
- İlaç toksisiteleri: Kardiyotoksisite (antrasiklin), mukozit, sitopeniler, TKİ sınıfına özgü yan etkiler vb.
Tedavi İlkeleri
1) İlaç Tedavileri
- Sitotoksik kemoterapi: Akut lösemilerde indüksiyon-konsolidasyon temeli.
- Hedefe yönelik ajanlar:
- TKİ’ler (KML ve Ph-pozitif ALL)
- FLT3, IDH1/2, BCL-2 (venetoklaks) inhibitörleri (AML alt grupları)
- Anti-CD20 (KLL), anti-CD38 (plazma hücreli olgular)
- Blinatumomab, inotuzumab (ALL)
- Diferansiyasyon tedavisi: All-trans retinoik asit (ATRA) ve arsenik trioksit (APL).
- Monoklonal antikor/konjugatlar ve hücresel tedaviler:
- CAR-T (özellikle CD19 hedefli, ALL/KLL belirli durumlar),
- Antikor-ilaç konjugatları (ADC).
2) İlaç Dışı Tedaviler
- Allojenik hematopoetik kök hücre nakli (allo-HKHN):
- Yüksek riskli, nüks veya MRD-pozitif akut lösemide küratif potansiyel.
- KML’de TKİ refrakter/ileri faz; KLL/AML’de seçilmiş olgular.
- Radyoterapi:
- Sınırlı kullanımlar: Kitle etkisi/kompresyon, SSS/testiküler tutulum, kemik ağrısı palyasyonu; nakil öncesi hazırlık rejimlerinde fraksiyonel TBI.
3) Destekleyici Tedavi ve Profilaksi
- Transfüzyon: Eritrosit ve trombosit; irradyasyon ve filtrasyon protokollerine dikkat.
- Antimikrobiyal profilaksi: Febril nötropeni kılavuzları doğrultusunda; Pnömosistis jirovecii profilaksisi uygun rejimlerde.
- Büyüme faktörleri: G-CSF seçilmiş durumlarda.
- Tümör lizisi önleme: Agresif hidrasyon, allopürinol veya rasburikaz.
- Koagülopati yönetimi: APL’de derhal ATRA başlanması ve hemostaz desteği.
- Destekleyici bakım: Beslenme, ağrı kontrolü, psikososyal destek, fertilite koruma stratejileri.
İzlem, Yanıt Değerlendirmesi ve Prognoz
- Hematolojik tam yanıt (CR): Kan değerlerinin düzelmesi ve kemik iliğinde blast baskılanması.
- MRD (asgarî rezidüel hastalık): Çok hassas akım sitometrisi veya moleküler yöntemlerle ölçülür; özellikle ALL ve AML’de tedavi uyarlamalarında belirleyicidir.
- Moleküler yanıt: KML’de BCR-ABL1 düzeyleri ile derin yanıt (MR^4-MR^4.5) tedavi hedeflerini tanımlar.
- Prognoz belirleyiciler: Yaş, performans durumu, tanı anı lökosit sayısı, sitogenetik/moleküler risk, tedaviye erken yanıt ve MRD durumu, komorbiditeler.
Ayırıcı Tanı
- Lökemid (leukemoid) reaksiyon: Reaktif lökositoz; toksik granülasyon ve olgun hücre hakimiyeti; klonal belirteç yoktur.
- Miyelodisplastik sendrom: Sitopeniler, displazi; blast eşiği ve klonal özellikler farklıdır.
- Lenfomaların lösemik fazı: İmmünfenotipleme ve genetik profil ayırır.
- Aplastik anemi: Pansitopeni + hiposellüler kemik iliği; blast artışı beklenmez.
Klinik Notlar ve Pratik İpuçları
- Acil durumlar: Ateşli nötropeni, leukostaz, APL-ilişkili DİK ve tümör lizisi kırmızı bayrak durumlardır; zamanında müdahale mortaliteyi belirgin azaltır.
- Fenotip-genotip eşleşmesi: Başlangıçta geniş panel moleküler testler, hedefe yönelik tedavi fırsatlarını belirler.
- MRD-yönelimli yaklaşım: Özellikle ALL ve AML’de nakil endikasyonu ve idame stratejileri için merkezî önemdedir.
- Kronik formlar: KLL’de tedavi başlama kriterleri (B semptomları, ilerleyici organomegali, hızla yükselen lenfositoz, sitopeniler) gözetilir; KML’de moleküler yanıt izlem programı standarttır.
Keşif
Aşağıdaki anlatı, löseminin iki yüzyıla yaklaşan serüvenini; ilk olgu betimlemelerinden sitogenetik devrimlere, kemoterapiden hedefe yönelik tedavilere ve hücresel immünoterapilere, nihayet güncel multi-omik ve ilaç geliştirme atılımlarına uzanan bir “bilimsel keşif hikâyesi” olarak bir araya getirir.
1) Erken izler: “beyaz kan” fikrinin belirişi (19. yüzyılın ilk yarısı)
- yüzyıl tıbbında mikroskopun rutinleşmesiyle kan, artık yalnızca kırmızı bir sıvı değil; hücresel bir evren olarak görülmeye başladı. 1820’ler–1840’larda Alfred Velpeau ve Alfred Donné’nin gözlemleri, olağandışı beyaz küre birikimlerine işaret ediyordu. 1845’te Edinburgh’da John Hughes Bennett, dalak ve karaciğer büyümesi eşliğinde kanda “irin benzeri” lökosit bolluğu bulunan bir olguyu yayımlayarak hastalığı “leucocythemia/beyaz hücreli kan” terimleriyle betimledi; iki yıl sonra Berlin’de Rudolf Virchow, aynı varoluşu “Leukämie/leukemia” adıyla andı ve tarihe hastalığın isim babası olarak geçti. Bu erken yıllar, löseminin bir “kanın dönüşümü” olduğu düşüncesini doğurdu; ne var ki kaynağının nerede olduğu belirsizdi.
2) Kanın beşiği bulunuyor: kemik iliği ve hücre sınıflaması (1868–1900)
1868’de Königsbergli patolog Ernst Neumann, postembriyonik yaşamda kan yapımının başlıca yerinin kemik iliği olduğunu göstererek hematopoezin “sahnesini” belirledi; ilerleyen yıllarda bu sahnenin başrolünde kök/öncül hücrelerin olduğu fikri güçlendi. Aynı yüzyılın son çeyreğinde Paul Ehrlich, anilin boyalarla geliştirdiği diferansiyel kan yayması ve granülosit/mast hücresi betimlemeleriyle lökosit alt tiplerini ayrıştırdı; bu, lösemilerin lenfoid ve miyeloid soylar olarak kavramsallaştırılmasının zeminini hazırladı.
3) Sitogenetik döneme giden yol ve “Philadelphia kromozomu” (1940’lar–1970’ler)
- yüzyılın ortası, lösemide iki büyük devrime sahne oldu: terapide folat antagonisti ile ilk remisyonlar ve kanserde tutarlı kromozom bozukluğunun keşfi. 1948’de Boston’da Sidney Farber, aminopterin ile çocukluk çağı akut lösemilerinde geçici de olsa dramatik remisyonlar bildirdi; modern kemoterapinin kapısı böyle aralandı.
1960’ta Philadelphia–Pennsylvania bağlantılı iki isim, Peter Nowell ve David Hungerford, KML’li hastalarda mikroskop altında “dakik (minute) bir kromozom” saptadı: Philadelphia kromozomu. Bu, insan kanserlerinde tutarlı bir kromozom anomalisinin ilk örneğiydi. 1973’te Chicago’dan Janet D. Rowley, yeni geliştirilmiş bantlama teknikleriyle bu yapının bir t(9;22) translokasyonu olduğunu gösterdi; ardından t(8;21) ve t(15;17) gibi başka imza translokasyonları da tanımladı. Sitogenetik bulguların, “kanser genetik bir hastalıktır” tezini kliniğe taşımasında Rowley’nin çalışmaları kurucu rol oynadı.
4) “Fikirden kür potansiyeline”: nakil, farklılaşma tedavisi ve hedefe yönelik devrim (1970’ler–2000’ler)
Farber’in açtığı yolda kombinasyon kemoterapileri gelişirken, E. Donnall Thomas’ın öncülüğünde allojenik kemik iliği/kök hücre nakli kavramı klinikte olgunlaştı; 1990’da bu çalışmalar Nobel ile onurlandırıldı. Leukemik klonu silip sağlıklı hematopoezle değiştirme fikri, yüksek riskli olgular için “kür potansiyeli” taşıyan bir paradigma sundu.
Bir diğer kırılma, akut promiyelositik lösemi (APL)’de gerçekleşti. 1980’lerden itibaren Şanghay ekolü (Zhen-Yi Wang, Zhu Chen ve çalışma arkadaşları), all-trans retinoik asit (ATRA) ile lösemik promiyelositleri farklılaştırarak hastalığı ölümcül DİK tablosundan “yüksek ölçüde kür edilebilir” bir duruma çevirdi; arsenik trioksit ile ATRA’nın akıllı kombinasyonları kemoterapisiz şemalara giden yolu açtı.
2001’de imatinib, BCR–ABL1 füzyon kinazını özgül olarak hedefleyerek KML’yi ölümcül bir hastalıktan yönetilebilir bir kronik duruma dönüştürdü; bu başarı, moleküler hedefli onkolojinin “kanıtı” oldu ve ikinci/üçüncü kuşak TKİ’lerle genişledi. Sonraki yıllarda asciminib, ABL’nin myristoyl cebine bağlanan ilk-in-sınıf STAMP inhibitörü olarak direnç paternlerini aşma stratejisini çeşitlendirdi.
5) Hücresel bağışıklığın sahneye çıkışı: CAR-T ve akıllı antikorlar (2010’lar–2020’ler)
2017’de FDA, çocuk ve genç erişkin B-ALL için ilk CAR-T ürününü (tisagenlecleucel) onayladı; bu karar, hastanın kendi T hücrelerinin yeniden programlanıp lösemik B hücrelerine yönlendirildiği bir çağın eşiğiydi. 2024’te CLL/SLL alanında da ilk CAR-T (lisocabtagene maraleucel/Breyanzi) hızlandırılmış onay aldı. Akıbetinde bispesifik T-hücre bağlayıcıları ve antikor-ilaç konjugatları, özellikle ALL ve KLL’de kemoterapi-dışı yaklaşımlara alan açtı.
6) Ölçebildiğin kadar kişiselleştir: MRD, paneller ve ELN çağının anatomisi (2010’lar–2020’ler)
Tedavi başarısı yalnız “tam remisyon”la değil, ölçülebilir artık hastalık (MRD) düzeyiyle de tanımlanır hâle geldi. Akım sitometrisi, PCR ve NGS tabanlı MRD testleri; ALL’de rutine girdi, AML’de prognostik ve tedavi uyarlama aracı olarak ağırlık kazandı. Paralelde, ELN 2022 gibi moleküler risk sınıflamaları (ör. NPM1, FLT3, TP53 vb.) endüksiyondan nakil kararına dek yol haritasını standartlaştırdı.
7) En güncel ufuk: menin ve ötesi (2024–2025)
2024’te FDA, KMT2A (MLL) translokasyonlu akut lösemilerde ilk menin inhibitörü revumenib’i onayladı; menin-KMT2A ekseninin farmakolojik baskılanması, özellikle pediatrik ve tedaviye dirençli alt gruplarda yeni bir hedefe-yönelik sayfa açtı. NPM1-mutant AML için de menin yolağını hedefleyen moleküller hızla ilerliyor. Aynı dönemde pirtobrutinib gibi kovalent olmayan (reversible) BTK inhibitörleri, KLL tedavi haritasını direnç mutasyonlarına rağmen genişletmeye başladı.
8) Haritanın “piksel çözünürlüğü”: tek hücreli ve bütünleşik çok-omik çağ
Bugün lösemiyi yalnız mutasyon listeleriyle değil, tek hücreli transkriptomik/epigenomik ve multi-omik atlaslarla, klonal evrim ve niche etkileşimini eşzamanlı okuyarak anlıyoruz. 2024–2025 arası çalışmalar, karma karyotipli AML’de tedaviye dirençli alt klonları ve hedeflenebilir fenotipleri tek hücre çözünürlüğünde ayırt etti; CML kök hücre altpopülasyonlarının TKİ’ye farklı yanıtlama kalıpları yine tek hücreli yaklaşımlarla çözümlendi. Bu veriler, MRD’nin yeniden tanımlanması, ilaç kombinasyonu tasarımı ve erken dönüş (early switch) kararlarını bilimsel zeminde keskinleştiriyor.
9) Hastalıktan önceki gölgeler: CHIP ve ön-lösemik durumlar
Genç bir kavram olan “belirsiz potansiyelli klonal hematopoez/CHIP”, görünürde sağlıklı bireylerde DNMT3A, TET2, ASXL1 gibi sürücü mutasyonları taşıyan kök hücre klonlarının yıllık yaklaşık %0,5–1 oranında hematolojik maligniteye evrilebildiğini gösterdi; aynı zamanda sistemik inflamasyon ve kardiyovasküler riskle de bağlantılı. Bu “ön sahne”, lösemi riskinin erken tanımlanması ve önleyici hematoloji için yeni sorular açıyor.
Bugünden yarına: nereye gidiyor?
Lösemi araştırmaları bugün üç geniş eksende derinleşiyor: (i) Kök-hücre biyolojisi ve klonal evrim (menin ekseni, menin-kombinasyonları ve NPM1-mutasyonlu AML hedefleri); (ii) Hücresel ve antikor-temelli immünoterapilerin erişkin AML ve KLL alt gruplarına, hatta kemoterapisiz protokollere doğru evrilmesi; (iii) Tek hücreli/bütünleşik multi-omik ile MRD ve riskin moleküler “yeniden kalibrasyonu”. Bu üç kulvarın kesiştiği yerde, tanı anından nükse, hatta ön-lösemik evrelerden tedavi-anahtarlama kararlarına kadar daha “ince ayarlı” bir kişiselleştirme ufku belirginleşiyor.
İleri Okuma
- Bennett, J. H. (1851). On Leucocythemia, or White Cell Blood. Edinburgh Medical and Surgical Journal, 76, 1–37. (PMC)
- Neumann, E. (1868). (Kan yapımında kemik iliğinin rolü üzerine orijinal gözlemler; tarihsel özet için) Translational Research Retrospective. (PMC)
- Ehrlich, P. (1879–1880). (Granülosit sınıflaması ve boyama teknikleri) Historical reviews. (ASM Journals)
- Farber, S. (1948). Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. New England Journal of Medicine, 238(23), 787–793. (New England Journal of Medicine)
- Nowell, P. C., & Hungerford, D. A. (1960). A minute chromosome in chronic granulocytic leukemia. Science (tarihsel anlatı ve anı yazıları için). (PMC)
- Rowley, J. D. (1977). 15/17 translocation, a consistent chromosomal change in APL. The Lancet, 1, 549–550. (Ayrıca 1973 t(9;22) için tarihsel kaynaklar). (PubMed)
- Zhou, G.-B., et al. (2005/2007). ATRA ve As2O3 ile APL farklılaşma tedavisi. Ann. Hematol.; PNAS. (PMC)
- Lasker Foundation (Druker, Lydon, Sawyers) (2009). CML’de hedefe yönelik tedaviye giden yol (imatinib). Lasker–DeBakey Award summary. (Lasker Foundation)
- FDA (2017). Tisagenlecleucel onayı (B-ALL). Drugs@FDA. (U.S. Food and Drug Administration)
- FDA (2021). Asciminib onayı (CML, STAMP). Drugs@FDA. (U.S. Food and Drug Administration)
- Döhner, H., et al. (2022). Diagnosis and management of AML in adults (ELN 2022). Blood, 140(12), 1345–1377. (ASH Publications)
- NCI / BMS (2024). CLL/SLL’de lisocabtagene maraleucel onayı. Kurumsal duyurular ve FDA sayfaları. (news.bms.com)
- FDA (2024). Revumenib (menin inhibitörü) onayı; KMT2A-düzenlemeli akut lösemi. Drugs@FDA. (U.S. Food and Drug Administration)
- Nature Genetics & eLife (2024). Tek hücreli/multi-omik ile AML/CML klonal evrim çalışmaları. Nat Genet; eLife. (Nature)
- Marnell, C. S., et al. (2021). CHIP ve lösemi riski. Journal of Molecular Biology (review). (PubMed)