
Muckle-Wells sendromu (MWS), kriyopirinle ilişkili periyodik sendromlar (CAPS) grubuna ait nadir bir otozomal dominant otoinflamatuar hastalıktır. Doğuştan gelen bağışıklık sisteminin düzenlenmesinde rol oynayan kriyopirin proteinini kodlayan NLRP3 genindeki (CIAS1 olarak da bilinir) mutasyonlardan kaynaklanır. Bu mutasyonlar tipik olarak proinflamatuar sitokin interlökin-1β’nin (IL-1β) aşırı üretimine yol açar. Klinik olarak, MWS cilt, eklemler ve diğer organ sistemlerini içeren tekrarlayan sistemik inflamasyon ataklarıyla karakterizedir. MWS’deki yaygın bir üçlü, epizodik ürtiker benzeri döküntüler, sensörinöral işitme kaybı ve amiloidoz gelişimini içerir; bunlardan ikincisi zamanla böbrek disfonksiyonuna yol açabilir.
İçindekiler
Genetik ve Patofizyoloji
NLRP3 geni kromozom 1’de (1q44) bulunur. Bu gendeki mutasyonlar normal inflamasom fonksiyonunu bozarak kaspaz-1’in kontrolsüz aktivasyonuna neden olur ve bu da IL-1β’nin aşırı üretimine yol açar. Bu proinflamatuar sitokin, MWS’de gözlemlenen ateşli ataklar, döküntü ve sistemik inflamasyondan birincil olarak sorumludur. Ek olarak, amiloidoza (özellikle AA amiloidozu) ilerleme, serum amiloid A proteininin çeşitli dokularda, özellikle böbreklerde birikmesiyle, inflamasyonun uzun vadeli yükünü yansıtır.
Klinik Sunum
- Epizodik ateşler ve döküntü: Hastalar genellikle ürtiker benzeri cilt lezyonlarıyla birlikte tekrarlayan ateşler yaşarlar. Bu ataklar stres, sıcaklık değişiklikleri veya diğer çevresel faktörler tarafından tetiklenebilir.
- Sensorinöral işitme kaybı: İlerleyen işitme bozukluğu, MWS’yi diğer kriyopirin ilişkili sendromlardan ayıran ayırt edici özelliklerden biridir. İşitme kaybının başlangıcı genellikle ergenlik veya erken yetişkinlikte görülür ve tedavi edilmezse ilerleyebilir.
- Artraljiler ve eklem ağrısı: Eklem semptomları genellikle iltihaplı alevlenmeler sırasında ortaya çıkar. Diğer romatolojik rahatsızlıklardan daha az şiddetli olsa da, tedavi edilmeyen iltihap yine de rahatsızlığa ve işlevsel bozukluğa yol açabilir.
- Böbrek tutulumu (amiloidoz): Sürekli iltihap, amiloid proteinlerinin (özellikle tip AA) birden fazla organda birikmesine neden olabilir ve sıklıkla böbrek fonksiyonunu etkiler. Zamanla, tedavi edilmezse kronik böbrek hastalığına yol açan proteinüri ve nefrotik sendrom gelişebilir.
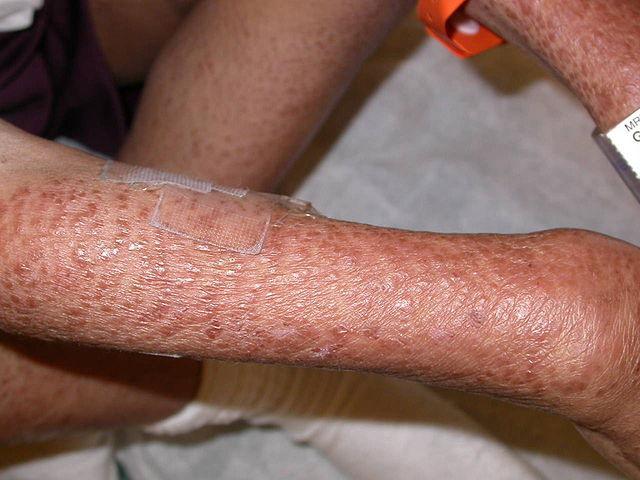
Tanı
MWS tanısı genellikle şunları içerir:
- Klinik değerlendirme: Epizodik ateşlere, döküntüye, işitme durumuna ve benzer semptomların aile geçmişine odaklanan ayrıntılı bir tıbbi geçmiş ve fizik muayene.
- Laboratuvar testleri: İnflamasyon belirteçleri (örn. C-reaktif protein ve serum amiloid A) alevlenmeler sırasında sıklıkla yükselir. Kalıcı yükselmeler semptomatik ataklar arasında bile devam eden inflamasyonu gösterir.
- Genetik test: NLRP3 genindeki mutasyonların tespiti tanıyı doğrular. Patojenik bir varyantın tanımlanması MWS’yi diğer otoinflamatuar sendromlardan ayırt etmeye yardımcı olabilir.
Yönetim ve Tedavi
- IL-1 inhibisyonu: Tedavinin temel dayanağı anakinra, canakinumab veya rilonacept gibi IL-1 blokaj terapilerini içerir. Bu ajanlar IL-1β aktivitesini hedef alarak sistemik inflamasyonu önemli ölçüde azaltır ve ilerleyici işitme kaybı ve amiloid birikimi gibi hastalık komplikasyonlarını önlemeye yardımcı olur.
- Amiloidoz için izleme: Amiloid birikiminden kaynaklanan böbrek bozukluğu riski göz önüne alındığında, proteinüri ve böbrek fonksiyonu için düzenli tarama hayati önem taşır.
- Destekleyici bakım: Stratejiler arasında eklem ağrısı ataklarının yönetimi, işitme keskinliğinin izlenmesi ve gerektiğinde işitme cihazları veya yardımcı dinleme cihazlarının düşünülmesi yer alır. Kapsamlı bakım için bir nefrolog, odyolog ve romatolog tarafından periyodik değerlendirmeler gerekebilir.
Prognoz
Erken tanı ve uygun IL-1 blokajı ile Muckle-Wells sendromunun uzun vadeli görünümü önemli ölçüde iyileşmiştir. Çoğu hasta, inflamatuar ataklarda belirgin bir azalma, işitme kaybı ilerlemesi riskinin azalması ve amiloidozla ilişkili komplikasyon olasılığının azalması yaşar. Bununla birlikte, devam eden gözetim ve tedaviye uyum, sürdürülebilir hastalık kontrolü için kritik öneme sahiptir.
Keşif
20. Yüzyılın Başından Ortalarına: Periyodik Sendromların Örnekleri
- 1900’lerin Başları: Çeşitli Avrupa tıp dergilerinde tekrarlayan ateşler, döküntüler ve iltihaplı ataklar açıklanmış olsa da bunlar henüz ayrı bir sendrom altında sınıflandırılmamıştır.
- 1950’ler: Doktorlar epizodik ürtiker, ateş ve işitme bozukluklarını olası kalıtsal veya ailevi durumlara bağlamaya başlamış ve gelecekteki adlandırma ve sınıflandırma için temel oluşturmuştur.
1962: Tom J. Muckle ve Michael Wells’in Resmi Açıklaması
- 1962: Tom J. Muckle ve Michael Wells, tekrarlayan ürtiker döküntüleri, ilerleyici sensörinöral işitme kaybı ve sistemik amiloidoz üçlüsüyle gelen hastaları tanımlayan, British Journal of Dermatology dergisinde çığır açan bir makale yayınladı. Bu üçlü, Muckle-Wells sendromu olarak bilinen şeyin klinik temel taşını oluşturdu.
1970’ler-1980’ler: Genişletilmiş Klinik İçgörüler ve Amiloidoz Tanıma
- 1970’ler: Daha fazla vaka çalışması, MWS’nin otozomal dominant kalıtım modelini vurgulayarak ailevi doğasını doğruladı. Romatologlar ve dermatologlar, ürtiker benzeri döküntüler, eklem ağrıları ve böbrek tutulumu (amiloidoz yoluyla) arasındaki ilişkileri giderek daha fazla fark ettiler. – 1980’ler: Amiloid birikimlerini tespit etme ve karakterize etme teknikleri iyileşti ve MWS hastalarında kalıcı inflamasyon ile amiloid birikimi arasında daha net bir korelasyon sağlandı. Araştırmacılar ve klinisyenler böbrek komplikasyonlarını geciktirmek veya önlemek için daha erken tanıya ihtiyaç olduğunu fark ettiler.
1990’lar: Otoinflamatuar Bir Durum Olarak Karakterizasyon
- 1990’ların Başından Ortalarına: Periyodik ateş sendromları üzerine devam eden araştırmalar, MWS’yi kalıtsal otoinflamatuar hastalıkların gelişen bir grubuna yerleştirdi. Artan farkındalık, MWS belirtilerinin genişliğini ve değişkenliğini anlamak için uluslararası kayıtları ve işbirlikli çalışmaları teşvik etti.
- 1990’ların Sonları: Peter N. Hawkins ve Ulusal Amiloidoz Merkezi’ndeki (UK) meslektaşlarının öncü çalışmaları, kronik inflamasyon, serum amiloid A (SAA) protein yükselmesi ve MWS’de amiloid birikimi arasındaki bağlantıyı daha da belirginleştirdi.
2000’lerin Başları: CAPS İçinde Genetik Atılımlar ve Sınıflandırma
- 2001–2002: Edward Aganna, Peter N. Hawkins, Seza Ozen ve diğerlerinin liderliğindeki çalışmalar, NLRP3 (aynı zamanda CIAS1 olarak da bilinir) genindeki mutasyonları belirleyerek, MWS, Ailevi Soğuk Otoinflamatuvar Sendrom (FCAS) ve Yenidoğan Başlangıçlı Multisistem İnflamatuvar Hastalık (NOMID) arasında paylaşılan genetik etiyolojiyi tanımladı. Bu üç durum birlikte, Kriyopirin İlişkili Periyodik Sendromlar (CAPS) olarak tanındı.
- 2003–2005: Sonraki araştırmalar, NLRP3 inflamazomunun rolünü ve interlökin-1β (IL-1β) düzenlemesini açıklığa kavuşturdu. Bu bulgular, araştırmayı IL-1 blokajını hedefleyen terapötik stratejilere yöneltti.
2000’lerin ortası–2010’lar: Hedeflenen IL-1 İnhibisyonunun Ortaya Çıkışı
- 2000’lerin ortası: Klinik deneyler, hiperinflamatuvar kaskadları kısıtlayarak MWS’de önemli semptom rahatlaması gösteren rekombinant bir IL-1 reseptör antagonisti olan anakinra’yı tanıttı. Bu, hastalık yönetiminde bir dönüm noktası oldu ve hastaların prognozunu değiştirdi.
- 2010’lar: Kanakinumab ve rilonacept gibi ek IL-1 hedefli tedavilerin geliştirilmesi ve onaylanması, sitokin inhibisyonunun terapötik vaadini vurguladı. İşitsel ve renal komplikasyonların erken ve etkili IL-1 blokajı ile önlenebilir veya kısmen geri döndürülebilir olduğu bulundu.
- 2010’ların sonu: Küresel kayıtlar ve çok merkezli çalışmalar, tekrarlayan inflamatuar ataklar ve sensörinöral işitme kaybı olan ailelerde erken genetik taramanın önemini vurgulayarak tanı kriterlerini iyileştirdi.
Mevcut ve Gelecekteki Yönler
- Devam eden araştırmalar, MWS’de genotip-fenotip korelasyonlarının anlaşılmasını genişletmeyi, anti-IL-1 tedavisinin uzun vadeli sonuçlarını iyileştirmeyi ve yeni biyolojik hedefleri araştırmayı amaçlamaktadır. Otoinflamatuar mekanizmalara ilişkin daha derin içgörüler ortaya çıktıkça, hassas, kişiselleştirilmiş tıp potansiyeli büyümeye devam etmektedir.
İleri Okuma
- Muckle, T.J., Wells, M. (1962). Urticaria, deafness, and amyloidosis. British Journal of Dermatology, 84(5), 419–427.
- Hawkins, P.N. (1998). Muckle-Wells syndrome: An autosomal dominant systemic amyloidosis. Quarterly Journal of Medicine, 91(3), 141–151.
- Aganna, E., Hawkins, P.N., Ozen, S., et al. (2002). Mutations in the NALP3/CIAS1/PYPAF1 gene in patients with Muckle–Wells syndrome and familial cold urticaria. Arthritis & Rheumatism, 46(8), 2189–2194.
- Lachmann, H.J., Paton, R., Bybee, A., et al. (2008). Clinical and genetic features of Muckle–Wells syndrome and related disorders of IL-1 β processing. Rheumatology, 47(3), 344–348.
- Kuemmerle-Deschner, J.B., Tyrrell, P.N., Koetter, I., et al. (2011). Efficacy and safety of anakinra therapy in pediatric and adult patients with the autoinflammatory Muckle–Wells syndrome. Arthritis & Rheumatism, 63(3), 840–849.