İçindekiler
Tanım ve Temel Kavramlar
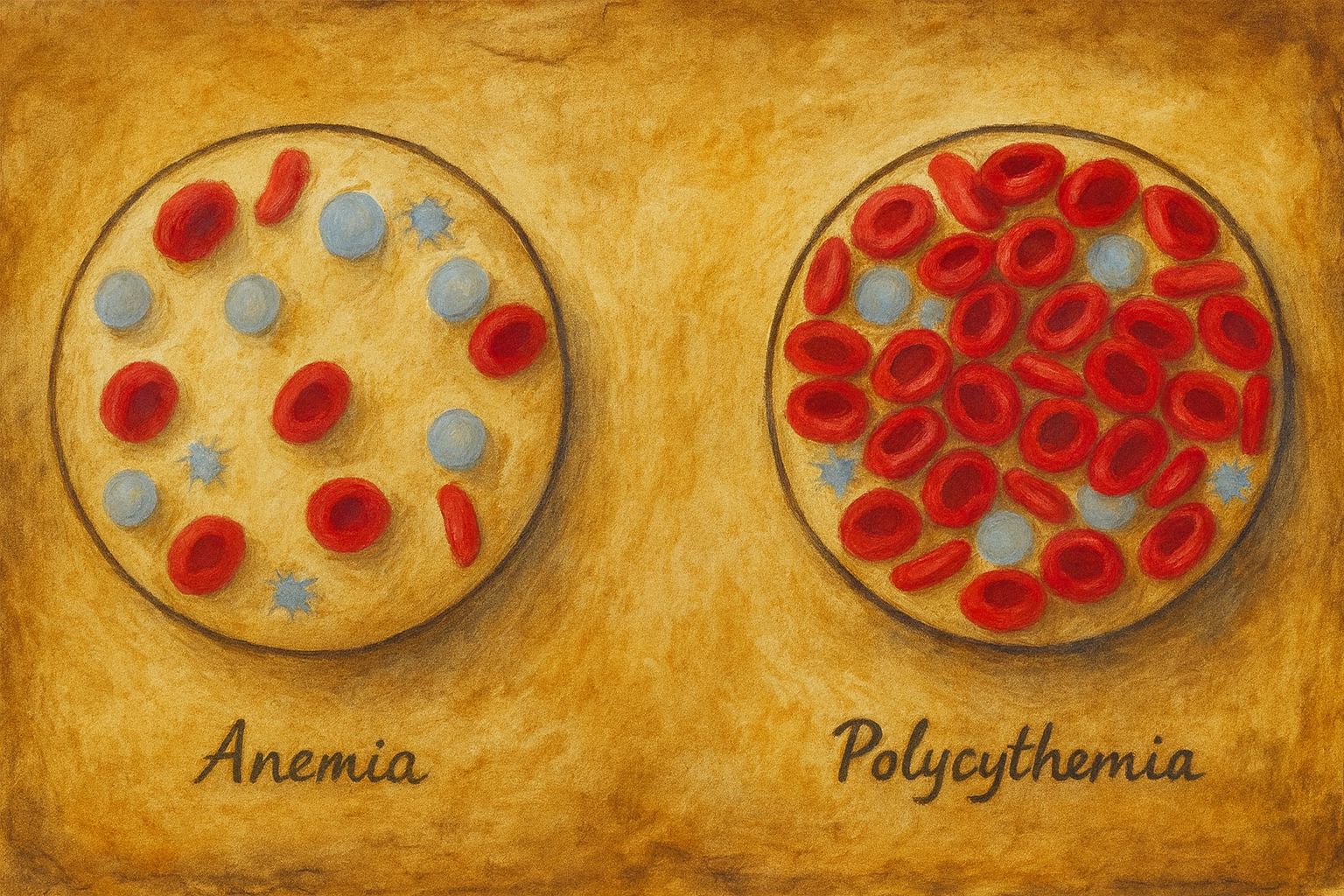
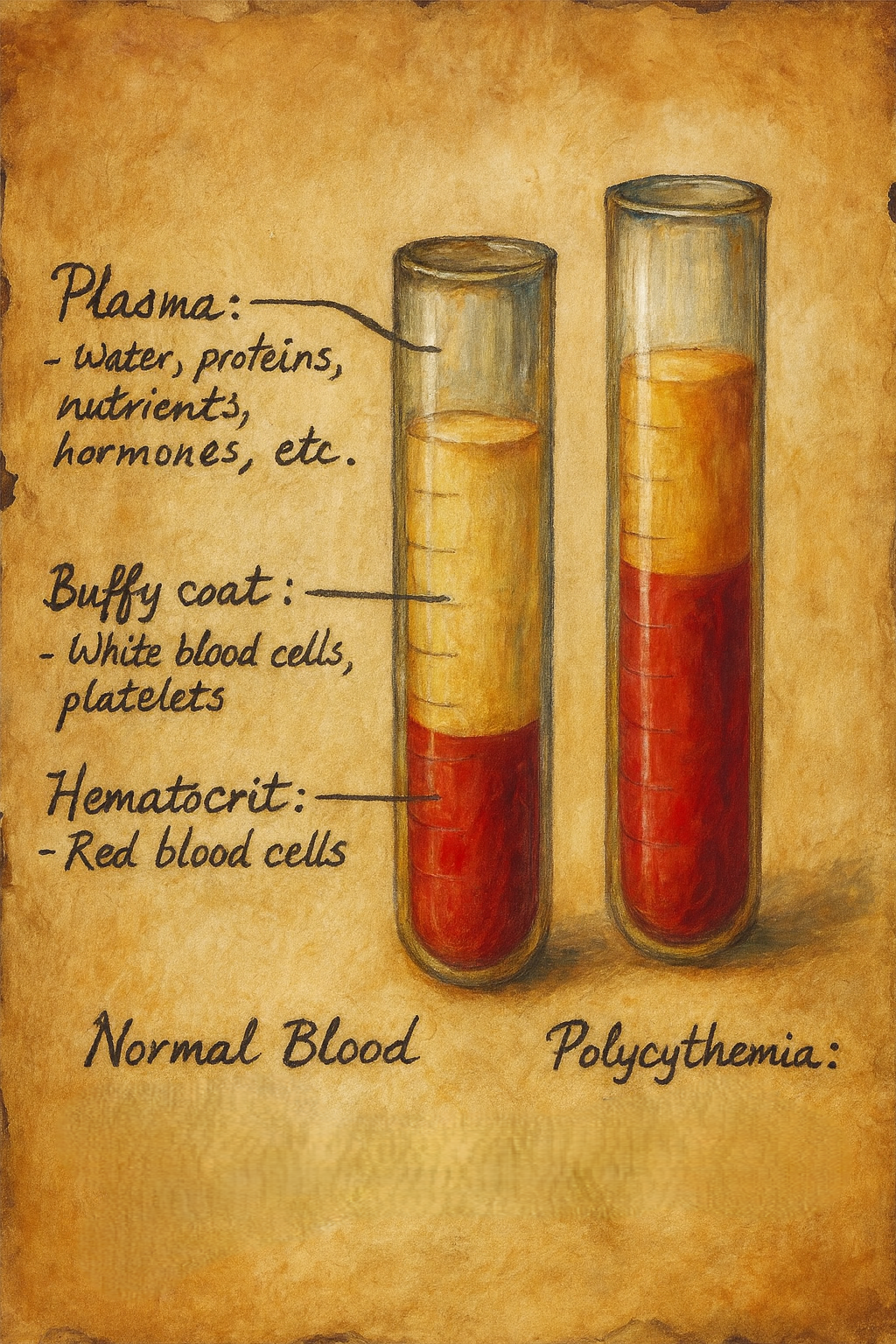
Polisitemi (klinik pratikte sıklıkla eritrositoz ile eş anlamlı kullanılır), dolaşımda eritrosit kütlesinin artışı ile karakterize bir tablodur. Laboratuvar düzeyinde bu artış, çoğunlukla hemoglobin (Hb) ve/veya hematokrit (Hct) yükselmesiyle ifade edilir. Güncel tanısal çerçeveler, erişkinlerde kabaca şu eşikleri dikkate alır:
- Erkek: Hb > 16,5 g/dL veya Hct > %49
- Kadın: Hb > 16,0 g/dL veya Hct > %48
Bu eşiklerin üzerinde persistan (kalıcı) bir yükseliş saptandığında, öncelikle artışın gerçek mi (mutlak eritrositoz) yoksa plazma hacmi azaldığı için göreli mi (relatif/apparent polisitemi) olduğunun ayırt edilmesi gerekir.
Polisitemi her zaman kanser midir?
Hayır. Polisitemi bir laboratuvar bulgusu/sendrom olup nedenine göre üç ana gruba ayrılır:
- Primer (klonal) eritrositoz/polisitemi: Polisitemia vera (PV) başta olmak üzere miyeloproliferatif neoplazi (MPN) grubunda yer alan kanser niteliğindeki hastalıklar.
- Sekonder eritrositoz/polisitemi: Doku hipoksisi veya eritropoetin (EPO) artışı ile ilişkili neoplastik olmayan nedenler (ör. KOAH, obstrüktif uyku apnesi, yüksek irtifa, EPO salgılayan tümörler, eksojen testosteron/EPO vb.).
- Relatif (apparent/stres) polisitemi: Plazma hacminin azalması (dehidratasyon, diüretik kullanımı, yoğun alkol/sigara, obeziteyle ilişkili Gaisböck sendromu) nedeniyle hematokritin göreli artışı; eritrosit kütlesi normaldir, kanser değildir.
Epidemiyoloji
Polisitemi, anemiye kıyasla belirgin derecede daha nadirdir. PV’nin yıllık insidansı düşük olup yaş ilerledikçe artar; tanı yaşı sıklıkla 60 yaş üzeri aralığındadır. Sekonder nedenler toplumda primer nedenlerden daha yaygındır (özellikle OSA, sigara, KOAH, yüksek irtifa maruziyeti).
Etiyoloji ve Patogenez
Primer: Polisitemia vera
PV, JAK-STAT yolunun uygunsuz aktivasyonu ile giden, JAK2 V617F (yaklaşık %95) veya JAK2 ekzon 12 (küçük bir alt grup) mutasyonlarıyla karakterize klonal bir miyeloproliferatif neoplazidir. Bu mutasyonlar EPO sin jaline bağımsız eritropoezi kolaylaştırır. PV’de çoğu zaman sadece eritrositler değil, lökosit ve trombositler de artar (pankitopeni değil, pansitofili).
Not: PV “kronik lösemi” olarak sınıflandırılmaz; miyeloproliferatif neoplazi başlığı altında yer alan kronik bir hematolojik malignitedir.
Sekonder eritrositoz: Hipoksi/EPO artışı
- Hipoksemi hipotezi: KOAH, interstisyel akciğer hastalığı, obstrüktif uyku apnesi, sağ-sol şantlı konjenital kalp hastalıkları, yüksek irtifa.
- Artmış EPO üretimi: Renal hücreli karsinom, hepatoselüler karsinom, hemangioblastom; renal hipoksi (renal arter stenozu, kistik böbrek hastalıkları).
- İlaçlar/ajanlar: Testosteron ve anabolik steroidler, dışarıdan EPO verilmesi; bazı hastalarda SGLT2 inhibitörleriyle belirgin hematokrit artışı bildirilebilir.
- Hemoglobin-oksijen afinitesi artışı: Yüksek afinite hemoglobin varyantları, 2,3-BPG eksikliği.
Relatif (apparent) polisitemi
Plazma hacmi azalır (dehidratasyon, diüretik kullanımı, aşırı alkol tüketimi, sigara, obezite/hiperadrenerjik durum); eritrosit kütlesi normal kalırken hematokrit göreli yükselir.
Ailesel/konjenital eritrositoz
Nadir mutasyonlar (örn. EPOR, VHL [Chuvash], EGLN1/PHD2, EPAS1/HIF-2α) kalıcı eritrositoza yol açabilir; bunlar kanser değildir.
Klinik Özellikler ve Komplikasyonlar
En sık erken belirti
PV ve diğer eritrositozlarda yorgunluk sık olmakla birlikte, PV için klasik ve ayırt ettirici erken belirti sıklıkla **“akvagenik pruritus”**tur (ılık/sıcak duş sonrası artan yaygın kaşıntı). Ayrıca:
- Baş ağrısı, baş dönmesi, bulanık görme, konsantrasyon güçlüğü
- Eritromelalji: Özellikle el ve ayaklarda yanma, kızarıklık ve şiddetli ağrı atakları
- Yüzde plethora, splenomegali (erken doyma, sol üst kadranda dolgunluk/ağrı)
- Hiperviskozite bulguları: Kulak çınlaması, görsel geçici ataklar, paresteziler
- Kanama (burun, diş eti) ve tromboz eğilimi bir arada olabilir; çok yüksek trombositlerde edinsel von Willebrand hastalığı gelişebilir
- Hiperürisemi ve gut atakları, ürik asit nefropatisi
Beyin ve sinir sistemi
Artmış viskozite serebral kan akımını azaltır; TIA/inme, baş ağrısı, bilişsel yavaşlama görülebilir. Neonatal dönemde uzamış polisitemi belirgin nörolojik riske yol açabilir; erişkinlerde de uzamış ve kontrolsüz hiperviskozite iskemik olay riskini artırır.
Tromboz ve kanama
PV’nin en ciddi sonuçları arteriyel/venöz trombozlardır. Budd–Chiari sendromu, portal/mezenterik ven trombozu, koroner/ serebral arteriyel olaylar görülebilir. Eşzamanlı kanama (özellikle mukozal) paradoksal değildir; trombosit işlev bozuklukları ve edinsel vWF eksikliği rol oynar.
Hastalığın seyri
PV yavaş seyirlidir. Uygun tedavi ile uzun yıllar stabil kalabilir. Zamanla post-PV miyelofibroza ve daha nadiren akut miyeloid lösemi (AML)’ye dönüşebilir. Literatürde AML dönüşüm riski düşük-orta düzeyde olup yıllar içinde kümülatif artış gösterir; risk, hasta yaşı, hastalık süresi, kullanılan sitoredüktif ajanlar ve moleküler yük ile ilişkilidir.
Tanısal Yaklaşım
1) Gerçek mi, göreli mi?
- Tekrarlayan CBC ile bulgunun kalıcı olduğu gösterilir.
- Klinik hidrasyon durumu, ilaçlar (özellikle diüretikler), sigara/alkol ve BMI değerlendirilir.
- Kuşku varsa eritrosit kütlesi ölçümü (nükleer tıp ile) relatif vs. mutlak ayrımını yapabilir; pratikte çoğu merkez laboratuvar ve klinik kombinasyonu ile karar verir.
2) Sekonder nedenler dışlanır mı?
- Oksijen satürasyonu (dinlenik ve gerekiyorsa gece/egzersiz), arter kan gazı; karboksihemoglobin (sigara/CO maruziyeti).
- EPO düzeyi: Sekonder nedenlerde sıklıkla yüksek/normal, PV’de genelde düşük.
- Akciğer fonksiyon testleri, polisomnografi (OSA kuşkusu), renal görüntüleme, kardiyak değerlendirme (şant), tümör taraması endikasyon dahilinde.
- P50 ölçümü ve gerekirse hemoglobin varyant analizi (yüksek afinite Hb şüphesinde).
3) Primer neden (PV) lehine mi?
- JAK2 V617F ve JAK2 ekzon 12 mutasyon analizi.
- Kemik iliği biyopsisi: Panmiyeloz (eritroid, granülositik ve megakaryositik hatlarda artış) ve tipik megakaryosit morfolojisi.
- Serum EPO genellikle düşük.
Güncel tanı çerçeveleri (WHO/ICC) PVSG’nin tarihsel kriterlerinin (örn. “eritrosit kütlesi artışı + normal O₂ satürasyonu + palpabl splenomegali”) yerini almıştır. Bugün majör kriterler (Hb/Hct yüksekliği, kemik iliği panmiyelozu, JAK2 mutasyonu) ve minör kriter (düşük serum EPO) üzerinden tanı konur. Eritrosit kütlesi ölçümü, eşik sınırında olanlarda yardımcıdır.
Laboratuvar ve Ek Bulgular
- CBC: Hb/Hct yüksek; lökositoz ve trombositoz PV’de sık.
- LDH, ürik asit yükselebilir; ferritin PV’de tekrarlayan flebotomilerle düşebilir (demir kısıtlı eritropoez).
- Periferik yayma: PV’de özgül olmayan; mikro/makro değişkenlik, trombosit kümeleri görülebilir.
- Koagülasyon: Çok yüksek trombositlerde ristosetin kofaktör aktivitesi/vWF antijeninde düşme (edinsel vWD) olabilir.
Ayırıcı Tanı
- Sekonder eritrositoz: Hipoksik akciğer/kalp hastalıkları, OSA, yüksek irtifa, sigara/CO, testosteron/EPO kullanımı, EPO üreten tümörler.
- Relatif polisitemi: Dehidratasyon, Gaisböck (obez/sigara/hipertansif erkeklerde sık).
- Konjenital eritrositoz: EPOR, VHL, EGLN1, EPAS1 mutasyonları; yüksek afinite hemoglobinler.
Tedavi İlkeleri
Tedavi hedefleri
- Tromboz riskini azaltmak (en kritik hedef).
- Hiperviskozite semptomlarını gidermek.
- Uzun vadede miyelofibroz/AML dönüşüm riskini en aza indirmek.
- Yaşam kalitesini artırmak (pruritus, eritromelalji, halsizlik).
Polisitemia vera yönetimi
- Flebotomi: En hızlı ve etkili ilk basamak. Hedef Hct < %45 (tüm erişkinler için güçlü kanıt). Gerektikçe tekrarlanır.
- Düşük doz aspirin (genellikle günde 75–100 mg): Mikrovasküler semptomlar ve tromboz profilaksisi için; kanama riski veya edinsel vWD varsa bireyselleştirme gerekir.
- Sitoredüktif tedavi (yüksek riskte):
- Yüksek risk tanımı sıklıkla ≥60 yaş veya geçirilmiş tromboz.
- Birinci seçenekler: Hidroksiüre (yaygın ve etkili) veya interferon-α türevleri (pegile/uzun etkili ropeginterferon; özellikle genç, gebelik planlayan ya da HU intoleransı olanlarda).
- İkinci basamak: Ruksolitinib (JAK1/2 inhibitörü), HU’ya dirençli/intoleran PV’de semptom kontrolü ve Hct hedefi için etkilidir.
- Diğer seçenekler (seçilmiş olgularda): Busulfan vb.
- Pruritus/Eritromelalji yönetimi: Aspirin, interferon veya ruxolitinib ile sıklıkla belirgin düzelme; semptomatik olarak soğuk duş, topikal ajanlar, antihistaminikler (sınırlı), SSRI/Gabapentin bazı olgularda yardımcı olabilir.
- Demir desteği: Flebotomi ile demir eksikliği gelişebilir; ancak demir replasmanı eritrositozu alevlendirebilir. Demir replasmanı ancak özel endikasyon ve yakın izlemle düşünülmelidir.
- Kardiyovasküler risklerin yönetimi: Hipertansiyon, dislipidemi, sigara bırakma, diyabet kontrolü, kilo yönetimi.
Sekonder eritrositoz yönetimi
- Nedene yönelik tedavi esastır:
- Hipoksemi: Oksijen tedavisi, altta yatan akciğer/kalp hastalığının optimizasyonu.
- OSA: CPAP tedavisi.
- Sigara: Bırakma programları; CO maruziyetinin kesilmesi.
- Testosteron/EPO: Doz gözden geçirilir, gerekirse azaltma/sonlandırma.
- Tümör: EPO üreten odakların tedavisi.
- Flebotomi: Hiperviskozite semptomu belirginse, cerrahi girişim öncesi ya da tromboz riski yüksek seçilmiş olgularda düşünülebilir; doku hipoksisini artırabileceği için dikkatli ve bireyselleştirilmiş kullanılmalıdır.
- Aspirin: Tromboz risk profiline göre düşünülür; kanama riskine duyarlı olunmalıdır.
Relatif polisitemi yönetimi
- Hidrasyonun düzeltilmesi, diüretik doz ayarı, alkol/sigara azaltımı/bırakma, kilo yönetimi ve altta yatan stresörlerin kontrolü ile Hct normalleşir. Sitoredüksiyon gerekmez.
Özel durumlar
- Gebelik: PV’de düşük doz aspirin yaygın; flebotomi ile Hct < %45 hedeflenir. Sitoredüktif ajan olarak interferon-α tercih edilir; hidroksiüre ve ruxolitinib gebelikte kullanılmaz.
- Cerrahi: Perioperatif dönemde tromboz/kanama riski yüksektir; Hct hedefe çekilmeli, antitrombotik strateji bireyselleştirilmelidir.
- Kaşıntı yönetimi: Ilık yerine serin duş, tahriş edici deterjanlardan kaçınma, fototerapi seçenekleri.
Sık Sorulan Sorulara Kısa Yanıtlar
- “Kırmızı kan hücresi sayımım yüksekse endişelenmeli miyim?”
Tek bir ölçümle hüküm verilmez. Yüksek irtifa, yoğun egzersiz, dehidratasyon, ilaçlar gibi fizyolojik/geçici nedenler olabilir. Tekrarlayan ölçüm ve klinik bağlam ile değerlendirilmelidir. - “Polisiteminin erken evresinde en sık belirti nedir?”
Yorgunluk sık olsa da PV için akvagenik pruritus daha karakteristiktir. Baş ağrısı, baş dönmesi, görme dalgalanmaları, eritromelalji de erken dönemde görülebilir. - “Polisitemi beyni etkiler mi?”
Evet. Hiperviskozite serebral perfüzyonu azaltır; baş ağrısı, TIA/inme riski artar. Kontrolsüz ve uzun süreli yükseklik en çok riski artırır. - “Polisitemi ailede görülür mü?”
PV olgularının büyük çoğunluğu kalıtsal değildir; somatik JAK2 mutasyonlarıyla edinilir. Nadir konjenital eritrositoz sendromları ailesel olabilir fakat bunlar PV’den ayrıdır. - “Polisiteminin en yaygın nedenleri nelerdir?”
Toplumda en sık sekonder ve relatif nedenler görülür: OSA, sigara/CO, KOAH, testosteron kullanımı; relatif grupta dehidratasyon, obezite, diüretikler, alkol. - “Polisitemi ne kadar hızlı ilerler?”
PV kronik ve yavaş seyirlidir; yıllar içinde komplikasyon veya dönüşüm gelişebilir. Uygun risk azaltımı ve tedaviyle uzun sağkalım mümkündür.
İzlem ve Prognoz
- Düzenli takip: CBC, semptom değerlendirmesi, tromboz/kanama öyküsü, Hct hedefi (<%45).
- Yan etkiler: Hidroksiüre ile sitopeni/deri değişiklikleri; interferonla flu-like belirtiler/otoimmünite; ruxolitinibte bağışıklık baskılanması riskleri izlenir.
- Sağkalım: Modern tedavilerle medyan sağkalım genel popülasyona göre kısalmış olsa da birçok seride yaklaşık 14–24 yıl bandında, genç/düşük riskli hastalarda daha uzundur. Esas belirleyiciler trombotik olayların kontrolü ve uygun risk yönetimidir.
Pratik Tanısal Akış (Özet Adımlar)
- Hb/Hct yüksekliği saptandı → yeniden ölç ve relatif nedenleri gözden geçir (hidrasyon, ilaçlar).
- Kalıcı yükseklik → SpO₂/ABG, karboksihemoglobin, EPO düzeyi.
- EPO düşük veya PV şüphesi → JAK2 (V617F ± ekzon 12), kemik iliği biyopsisi.
- EPO yüksek/normal → Akciğer/kalp değerlendirmesi, OSA için tarama, renal görüntüleme; gerektiğinde hemoglobin afinitesi/P50.
- Bulgulara göre PV / sekonder / relatif ayrımı ve hedefe yönelik tedavi.
Keşif
Tıbbın 19. yüzyılın sonundaki laboratuvar devriminde, mikroskobun altında görünen her hücre, hastalığı bir anda “fikir” olmaktan çıkarıp ölçülebilir bir varlık hâline getiriyordu. O günlerde Paris’te çalışan Louis Henri Vaquez, 1892’de siyanoz, dalak büyüklüğü ve olağandışı eritrositozun bir arada seyrettiği dikkat çekici bir olguyu yayımladı; sıradan bir “plethora” değil, kendine özgü bir kan hastalığı olduğuna inanıyordu. Bu gözlem kısa sürede yankı buldu: 1903’te William Osler, aynı tabloyu Kuzey Amerika’da sistematik biçimde tanımladı ve iki isim zamanla birlikte anıldı—maladie de Vaquez, Vaquez–Osler hastalığı ya da modern adıyla polisitemia vera (PV). Bu erken anlatılar, hastalığın damar içi “yoğunluk” metafiziğinden, klinik bir varlık olarak eritrosit kütlesi artışına doğru taşınmasında tarihsel mihenklerdi.
1951’e gelindiğinde, Boston’dan William Dameshek, birbirine akraba görünen birkaç kronik kan hastalığını tek bir şemsiyede toplama cesareti gösterdi: miyeloproliferatif hastalıklar. PV’yi, esansiyel trombositemi (ET), primer miyelofibroz (PMF) ve o günün dilindeki diğer “miyeloproliferatif” birlikteliklerle aynı soydan kabul etti. Dameshek’in sezgisi, sonraki on yıllarda hem tanısal ölçütlere hem de araştırma gündemine yön verdi; PV artık yalnız bir “fazlalık” değil, kök hücre temelli klonal bir süreçti.
1967’de kurulan Polycythemia Vera Study Group (PVSG), PV’nin hem tanısını standardize etti hem de tedavide ilk randomize adımları attı. Klasik PVSG kriterleri—artmış eritrosit kütlesi, yeterli oksijen satürasyonu, dalak büyüklüğü—bir kuşağın diline yerleşti. Tedavi cephesindeyse flebotominin karşısına dönemin “rasyonel” miyelosupresifleri olan P³² ve klorambusil çıkarıldı; zaman, bu ajanların lökomojenik maliyetlerini öğretti ve flebotomi kolunun görece daha iyi sağkalımı tarih sayfalarına not edildi. 1970’ler ve 80’ler, hidroksiürenin daha az mutajenik bir seçenek olarak öne çıktığı yıllardı; PV’nin tedavi tarihi, bilimin etki kadar bedel hesabını da birlikte yapmayı öğrendiği bir “uzun prova” gibiydi.
Moleküler çağ 2005’te birden hızlandı: Birkaç bağımsız grup, PV olgularının büyük kısmında JAK2 V617F mutasyonunu gösterdi; JAK-STAT yolunun otonom ateşlenmesi, eritropoezi EPO sinyalinin gölgesinden çıkarıyor, klonal bir dönüşümün kapısını aralıyordu. Bir yıl sonra, bu mutasyonun canlı modellerde PV-benzeri fenotipi başlatabildiği gösterildi; moleküler bulgu, neden ile görüntü arasındaki boşluğu kapatan bir köprüye dönüştü. 2009’dan itibaren JAK2 46/1 germline haplotipinin MPN’lere yatkınlığı artırdığına dair veriler geldi; topluca okunduğunda, PV’nin hikâyesi basit bir “çok hücre üretimi” anlatısından, genetik yatkınlık + edinilmiş sürücü mutasyon diyalektiğine evrildi.
Tanı ölçütleri de bu moleküler berraklığa göre yeniden yazıldı. WHO 2016 revizyonu hemoglobin/hematokrit eşiklerini klinik gerçekliğe yaklaştırdı; JAK2 varlığı, kemik iliği panmiyelozu ve düşük EPO gibi sütunlarla PV’yi sekonder eritrositozdan ayıran çerçeveyi güçlendirdi. 2022 ve 2025 değerlendirmeleri, JAK2 mutasyonu ile Hb/Hct düzeylerinin birlikteliğini merkezine alan güncel bir dil benimsedi. RBC kütlesi ölçümü artık sınırda olgular için yardımcı, fakat çoğu vakada genetik ve histomorfoloji ile yerini bulmuş durumda.
Tedavide dönüm noktaları ise kanıt temelli birkaç taşla örüldü. ECLAP çalışması (2004), düşük doz aspirinin trombotik olayları güvenle azalttığını gösterdi; CYTO-PV (2013), hematokrit hedefini <%45 belirlemenin kardiyovasküler ölüm ve majör trombozu anlamlı biçimde düşürdüğünü kanıtladı. Hidroksiüre, özellikle yüksek risklilerde omurgayı oluşturmayı sürdürürken—intolerans/dirençte—ruksolitinib (JAK1/2 inhibitörü) PV’de hematokrit kontrolü, dalak ve semptom yükünde üstünlük sağlayarak ikinci basamakta yeni bir kapı araladı. Aynı dönemde interferon-α ailesi, özellikle ropeginterferon alfa-2b ile uzun erimli hem hematolojik hem moleküler yanıtların mümkün olabileceğini göstererek “hastalık değiştirici” potansiyele dair kuvvetli bir ipucu sundu.
Polisiteminin hikâyesi yalnızca PV ile sınırlı değil; eritrositoz evreninin kenar mahallelerinde kalıtsal yol ayrımları da var. EPOR genindeki germline durdurucu mutasyonlar primer ailesel/konjenital eritrositozu doğururken, oksijen algılama (HIF) eksenindeki VHL/EGLN1/EPAS1 mutasyonları sekonder ama kalıtsal eritrositoz tablolarına yol açıyor. Volga kıyılarından İtalya’nın Ischia adasına uzanan Chuvash eritrositozu öyküsü, VHL’deki R200W varyantının “hipoksi programı”nı paradoksal biçimde kalıcı açık konumuna getirdiğini öğretti. Bu damar, PV’nin ne olmadığını aydınlatırken, eritrositozun tüm biçimlerini sistem biyolojisi penceresinden yeniden okumamıza yardım etti.
Bugünün klinik pratiğinde PV yönetimi; flebotomi ile Hct’yi <%45’te tutmak, düşük doz aspirin, sitoredüksiyon gereksinimini yaş ve tromboz öyküsüne göre katmanlamak ve gerektiğinde interferon veya ruksolitinibe başvurmak üzerine kurulu. Ancak güncel araştırma, çıtayı daha yükseğe koyuyor: JAK2 alel yükünün uzun erimli gidişatla ilişkisi, 46/1 haplotipinin üç boyutlu kromatin mimarisinde MPN başlatıcı riskini nasıl şekillendirdiği, interferonların klonu geriletme kapasitesinin miyelofibroza dönüşüm riskine etkisi ve “masked”/erken PV evrelerinde optimal müdahale zamanı… Hepsi, bundan sonraki bölümün başlıkları. Klinik bağlamda ELN ve diğer grupların 2021–2024 önerileri, düşük riskte “flebotomi + aspirin” omurgasını korurken; sitoredüksiyon endikasyonlarını tromboz riski, semptom yükü, dalak ve hematokrit kontrolündeki güçlükler üzerinden daha incelikli bir dile çeviriyor.
Ve nihayet, hikâyenin başlangıçtaki ilk sahnesine dönelim: Vaquez’in koyu kırmızı yüzlü hastası ile Osler’in ayrıntılı vakaları, bizi eritrosit kütlesini ölçmeyi öğrenmeye mecbur bırakmıştı. Bugün ise aynı sahneye, JAK2’nin kodon 617’si, kemik iliğinin megakaryosit morfolojisi, EPO düzeyi ve klonal varyant yükü eşlik ediyor. Yol boyunca PVSG’nin uyardığı her ihtiyat, ECLAP’ın getirdiği her aspirin tableti, CYTO-PV’nin çizdiği her %45 sınırı ve interferonun telkin ettiği her moleküler sessizlik—hepsi bu uzun öykünün satır aralarında. Polisiteminin tarihi, aslında bir yakınlaştırma tarihidir: makroskopikten mikroskopiğe, klinikten genoma, anekdottan deneye… Daha ince gördükçe daha emin tedbirler, daha hedefli tedaviler ve giderek daha kişisel bir kan hekimliği.
İleri Okuma
- Vaquez, L.H. (1892). Sur une forme spéciale de cyanose s’accompagnant d’hyperglobulie excessive et persistante. Comptes Rendus des Séances de la Société de Biologie et de ses Filiales, 44, 384–388.
- Osler, W. (1903). Chronic cyanosis, with polycythemia and enlarged spleen: A new clinical entity. American Journal of the Medical Sciences, 126(1), 187–201.
- Dameshek, W. (1951). Some speculations on the myeloproliferative syndromes. Blood, 6(4), 372–375.
- Berlin, N.I. (1975). Diagnosis and classification of the polycythemias. Seminars in Hematology, 12(4), 339–351.
- Wasserman, L.R., Berk, P.D., & Berlin, N.I. (1979). Polycythemia Vera and the Polycythemia Vera Study Group. Seminars in Hematology, 16(4), 159–182.
- Pearson, T.C. (1981). Apparent and true polycythaemia: Definition, prevalence and causes. British Journal of Haematology, 48(4), 557–567.
- Spivak, J.L., & Silver, R.T. (1987). The polycythemias: Polycythemia vera and secondary polycythemia. Current Problems in Cancer, 11(3), 67–122.
- Adamson, J.W., & Fialkow, P.J. (1978). Clonal analysis of polycythemia vera: A stem-cell disease. New England Journal of Medicine, 299(13), 688–694.
- Baxter, E.J., Scott, L.M., Campbell, P.J., East, C., Fourouclas, N., Swanton, S., … & Green, A.R. (2005). Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet, 365(9464), 1054–1061.
- Kralovics, R., Passamonti, F., Buser, A.S., Teo, S.S., Tiedt, R., Passweg, J.R., … & Skoda, R.C. (2005). A gain-of-function mutation of JAK2 in myeloproliferative disorders. New England Journal of Medicine, 352(17), 1779–1790.
- James, C., Ugo, V., Le Couédic, J.P., Staerk, J., Delhommeau, F., Lacout, C., … & Vainchenker, W. (2005). A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature, 434(7037), 1144–1148.
- Levine, R.L., Wadleigh, M., Cools, J., Ebert, B.L., Wernig, G., Huntly, B.J.P., … & Gilliland, D.G. (2005). Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell, 7(4), 387–397.
- Percy, M.J., Furlow, P.W., Lucas, G.S., Li, X., Lappin, T.R., McMullin, M.F., & Lee, F.S. (2008). A gain-of-function mutation in the HIF2A gene in familial erythrocytosis. New England Journal of Medicine, 358(2), 162–168.
- McMullin, M.F., Reilly, J.T., Campbell, P., Bareford, D., Green, A.R., Harrison, C.N., … & Mead, A.J. (2016). Guideline for the diagnosis, investigation and management of polycythaemia/erythrocytosis. British Journal of Haematology, 172(3), 422–432.
- Marchioli, R., Finazzi, G., Landolfi, R., Kutti, J., Gisslinger, H., Patrono, C., … & Barbui, T. (2004). Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. Journal of Clinical Oncology, 23(10), 2224–2232.
- Marchioli, R., Finazzi, G., Specchia, G., Cacciola, R., Cavazzina, R., Cilloni, D., … & Barbui, T. (2013). Cytoreductive therapy in polycythemia vera: Results of the CYTO-PV randomized clinical trial. New England Journal of Medicine, 368(1), 22–33.
- Barbui, T., Finazzi, G., Carobbio, A., Thiele, J., Passamonti, F., Rumi, E., … & Tefferi, A. (2015). Development and validation of an International Prognostic Score of thrombosis in World Health Organization–essential thrombocythemia (IPSET-thrombosis). Blood, 120(26), 5128–5133.
- Gisslinger, H., Klade, C., Georgiev, P., Kroeger, N., Gercheva-Kyuchukova, L., Egyed, M., … & Silver, R.T. (2020). Ropeginterferon alfa-2b versus standard therapy for polycythemia vera (PROUD-PV and CONTINUATION-PV): A randomized, non-inferiority, phase 3 trial. Lancet Haematology, 7(3), e196–e208.
- Tefferi, A., & Barbui, T. (2023). Polycythemia vera: 2023 update on diagnosis, risk-stratification and management. American Journal of Hematology, 98(1), 41–58.
- Spivak, J.L. (2024). Contemporary understanding of polycythemia vera pathogenesis and therapy. Blood Reviews, 58, 101124.