
Rett sendromu, öncelikle kadınları etkileyen ve X kromozomundaki MECP2 genindeki mutasyonlarla bağlantılı olan nadir, ilerleyici bir nörogelişimsel bozukluktur. Yaygın gelişimsel bir bozukluk olarak ICD-10 kodu F84.2 altında sınıflandırılır. Aşağıda, Rett sendromunun mevcut anlayışının, epidemiyolojisinin, nedenlerinin, genetiğinin, klinik sunumunun, ilerlemesinin ve yönetiminin ayrıntılı bir dökümü, ek klinik bilgilere dayanarak çapraz kontrol edilmiş ve genişletilmiştir.

Epidemiyoloji:
- Rett sendromunun prevalansı yaklaşık olarak 10.000 ila 15.000 kadında 1’dir, ancak bazı kaynaklar 22.800’de 1 kadar düşük oranlar bildirmektedir. Bu tutarsızlık, tanı kriterlerindeki değişkenlik ve özellikle daha hafif veya atipik vakalarda eksik raporlama nedeniyle ortaya çıkmaktadır.
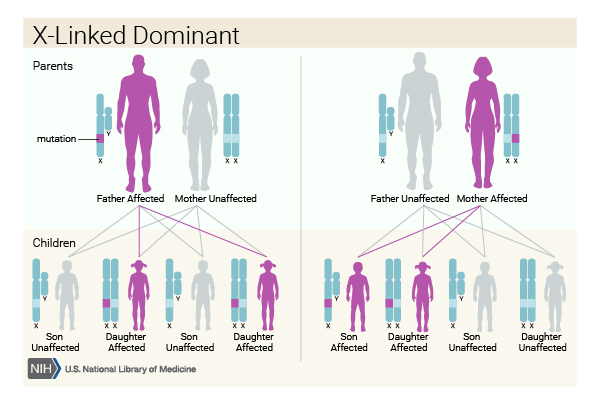
- Rett sendromu, X’e bağlı dominant kalıtım modeli nedeniyle ağırlıklı olarak kız çocuklarını etkiler. Bozukluk, tek bir X kromozomunda meydana geldiğinde mutasyonun etkilerinin ciddiyeti nedeniyle tipik olarak hayatta kalamayan erkek çocuklarda çok nadir görülür. Erkek vakaları son derece nadirdir ve genellikle somatik mozaiklik veya Klinefelter sendromu (47,XXY) içerir.
Tarihsel Arka Plan:
Andreas Rett tarafından ilk açıklama (1966):
- Avusturyalı çocuk doktoru Andreas Rett** sendromu ilk kez 1966 yılında tanımlamıştır. Bir grup kız çocuğunda gelişimsel gerileme, motor bozukluk ve karakteristik tekrarlayan el hareketleri gözlemlemiş ve bulgularını belgelemiştir. Bununla birlikte, çalışması nispeten belirsiz bir dergide Almanca olarak yayınlandı ve bu da daha geniş bilimsel topluluktaki ani etkisini sınırladı.
- Rett, el fonksiyon kaybı, “el yıkama” hareketleri, motor bozulma ve entelektüel gerileme gibi ayırt edici semptomları tanımlamıştır.

Bengt Hagberg (1983) tarafından **Uluslararası Tanınma:
- İsveçli çocuk doktoru Bengt Hagberg** 1983 yılında bu duruma uluslararası dikkat çekmiştir. Hagberg, meslektaşlarıyla birlikte, benzer semptomlar sergileyen 35 kızdan oluşan bir grubu analiz ederek Rett sendromunu ayrıntılı olarak tanımlayan ilk İngilizce makaleyi yayınladı.
- Hagberg’in yayını, Andreas Rett’in ardından bu durumu resmi olarak “Rett sendromu” olarak adlandıran ve tıp dünyasında geniş çapta tanınmasını sağlayan ilk yayın oldu. “Otizm, demans, ataksi ve kız çocuklarında amaca yönelik el kullanımının kaybından oluşan ilerleyici bir sendrom: Rett sendromu” başlıklı makale, sendromun anlaşılmasında bir dönüm noktası olmuştur.
Tanı Kriterlerinin Oluşturulması (1980’ler-1990’lar):
- 1980’ler ve 1990’lar** boyunca, araştırmacılar Rett sendromu için teşhis kriterlerini geliştirmeye başladılar. Bu zamana kadar, otizm spektrum bozuklukları kategorisi altında bir pervasif gelişimsel bozukluk olarak sınıflandırıldı (ICD-10: F84.2).
- Pediatristler ve nörologlar, Rett sendromunu diğer durumlardan ayıran karakteristik gelişimsel gerileme, motor disfonksiyon ve otistik benzeri davranışları tanımladılar.
MECP2 Gen Mutasyonunun Tanımlanması (1999):
- En önemli atılım 1999 yılında, Lübnan asıllı Amerikalı bir doktor ve genetikçi olan Huda Zoghbi, X kromozomu üzerindeki MECP2 genindeki mutasyonların Rett sendromu vakalarının çoğundan sorumlu olduğunu keşfettiğinde gerçekleşti. Bu keşif Nature dergisinde yayınlandı ve bozukluğun genetik temelinin anlaşılmasını değiştirdi.
- MECP2 (metil-CpG bağlayıcı protein 2)** beyin gelişiminde rol oynayan diğer genleri düzenleyen kritik bir gendir. MECP2’deki mutasyonlar normal nöronal işlevi bozarak Rett sendromunda görülen ilerleyici nörolojik gerilemeye yol açar.
Erkek Vakaları ve MECP2 Duplikasyonunu Anlamak (2000’lerin Başı):
- 2000’lerin başında**, daha fazla araştırma erkeklerde nadir görülen *Rett sendromu* vakalarını araştırdı ve MECP2 mutasyonları olan çoğu erkek hastanın bebeklik döneminde hayatta kalamadığını, bazılarının ise somatik mozaiklik veya Klinefelter sendromu gibi diğer kromozomal anormalliklerle hayatta kaldığını açıkladı.
- Ayrıca araştırmacılar, erkek çocuklarda Rett benzeri bir fenotiple sonuçlanan ve MECP2 geninin fazladan kopyalarını içeren bir durum olan MECP2 duplikasyon sendromunu keşfetmişlerdir.
Ek Genetik Nedenlerin Keşfi (CDKL5 ve FOXG1 Genleri, 2004-2010’lar):
- Çoğu Rett sendromu vakası MECP2’deki mutasyonlarla bağlantılı olsa da, daha sonraki çalışmalar CDKL5 (sikline bağımlı kinaz benzeri 5) ve FOXG1 (Forkhead box protein G1) gibi diğer genlerde atipik Rett sendromuna veya Rett benzeri semptomlara neden olabilen mutasyonları tanımlamıştır. Bu keşifler, bozukluğun genetik heterojenliğini vurgulamıştır.
- CDKL5 mutasyonları** daha erken nöbet başlangıcı ve daha ciddi gelişimsel gecikme ile ilişkiliyken, FOXG1 mutasyonları gelişimsel sorunların doğumdan itibaren belirgin olduğu konjenital Rett sendromu vakalarında yer almaktadır.
Hayvan Modellerinin Geliştirilmesi (2000’ler-2010’lar):
- MECP2 mutasyonunun keşfinin ardından araştırmacılar, bozukluğun patolojisini incelemek ve potansiyel tedavileri test etmek için Rett sendromunun fare modellerini geliştirdiler. Bu modeller, Rett sendromunun nörolojik semptomlarını taklit ederek hastalığın ilerleyişine dair içgörü sağlamıştır.
- Bu hayvan modelleri, klinik öncesi araştırmaların ilerletilmesinde etkili olmuş ve gen terapisi ve farmakolojik müdahalelerin test edilmesi için temel oluşturmuştur.
Gen Terapisi ve Farmakolojik Denemeler (2010’lar-Günümüz):
- Son yıllarda, gen terapisi Rett sendromunu tedavi etmek için umut verici bir yol olarak ortaya çıkmıştır. Bilim insanları, kusurlu MECP2 genini değiştirmenin veya onarmanın yollarını araştırmaktadır. Hayvan modellerinde gen terapisi kullanan erken preklinik çalışmalar, Rett benzeri semptomları tersine çevirme potansiyelini göstermiştir.
- gibi ilaçlar için klinik denemeler
Gen Tedavisi ve Farmakolojik Denemeler (2010’lar-Günümüz):
- Son yıllarda, gen terapisi Rett sendromunun tedavisi için umut verici bir yol olarak ortaya çıkmıştır. Bilim insanları, kusurlu MECP2 genini değiştirmenin veya onarmanın yollarını araştırmaktadır. Hayvan modellerinde gen terapisi kullanan erken klinik öncesi çalışmalar, Rett benzeri semptomları tersine çevirme potansiyelini göstermiştir.
- Rett sendromu hastalarında apne gibi solunum sorunlarını tedavi etmeyi amaçlayan sarizotan gibi ilaçlar ve MECP2 eksikliğinin aşağı akış etkilerini hedefleyen diğer bileşikler için klinik denemeler halen devam etmektedir.
Uluslararası Rett Sendromu Vakfı ve Savunuculuk (2000’ler-Günümüz):
- Uluslararası Rett Sendromu Vakfı (IRSF)** gibi kuruluşların kurulması, farkındalığın artırılmasında, araştırmaların finanse edilmesinde ve Rett sendromundan etkilenen ailelerin desteklenmesinde çok önemli bir rol oynamıştır.
- Küresel farkındalık kampanyaları ve hasta savunuculuğu, bilim insanları, bakıcılar ve klinisyenler arasında araştırma finansmanının ve işbirliğinin artmasına yol açarak Rett sendromu tedavilerine yönelik araştırmaları ilerletmiştir.
Nedenleri:
- Vakaların % 90’ından fazlasında Rett sendromuna MECP2 (metil-CpG bağlayıcı protein 2) genindeki mutasyonlar neden olur. MECP2, metillenmiş DNA’ya bağlanarak ve gen ekspresyonunu kontrol ederek diğer genlerin düzenlenmesinde rol oynadığı için nöronal fonksiyon için çok önemlidir.
- MECP2 duplikasyonları** Rett sendromu vakalarının küçük bir alt kümesini oluşturur. Nadir durumlarda, CDKL5 (siklin bağımlı kinaz benzeri 5) veya FOXG1 (Forkhead box protein G1) gibi diğer genlerdeki mutasyonlar Rett benzeri semptomlarla ilişkilidir.
- Bu mutasyonlar tipik olarak de novo yani sperm veya yumurta oluşumu sırasında kendiliğinden meydana gelir ve ebeveynlerden miras alınmaz.
Genetik Kalıtım:
- Rett sendromu X’e bağlı baskın kalıtım modelini takip eder. Çoğu durumda, etkilenen bireyler mutasyonu ebeveynlerinden ziyade de novo olarak miras alırlar.
- Kadınlar** tipik olarak, normal X kromozomu tarafından bir miktar telafiye izin veren iki X kromozomunun varlığı nedeniyle etkilenir. Erkeklerde** sadece bir X kromozomuna sahip olmak, MECP2 mutasyonunun ciddi gelişimsel sorunlara neden olabileceği ve genellikle doğum öncesi veya doğum sonrası erken ölüme yol açabileceği anlamına gelir. Bununla birlikte, tipik olarak somatik mozaizm (sadece bazı hücrelerin mutasyonu taşıdığı) veya Klinefelter sendromunda (47,XXY) görüldüğü gibi ekstra bir X kromozomu içeren nadir Rett sendromlu erkek vakaları mevcuttur.
Semptomlar:
Rett sendromu, tipik olarak gelişimin çeşitli aşamalarında ilerleyen çok çeşitli nörolojik ve motor semptomlara sahiptir. Temel özellikler şunları içerir:
- El stereotipleri, özellikle karakteristik ve tanısal olan tekrarlayan el sıkma veya “yıkama” hareketleri.
- Amaca yönelik el kullanımı, konuşma ve motor beceriler dahil olmak üzere edinilmiş becerilerin** kaybı.
- Sosyal etkileşime olan ilgi genellikle devam etse de, sosyal geri çekilme ve göz temasının azalması gibi Otistik benzeri davranışlar.
- Ciddi bilişsel bozukluk**: Entelektüel gelişim önemli ölçüde bozulur ve bunamaya benzer bir duruma yol açar.
- Mikrosefali (baş büyümesinin azalması), görünüşte normal bir gelişim döneminden sonra fark edilir hale gelir.
- Nöbetler**: Rett sendromlu birçok çocukta *epilepsi* gelişir.
- Spastisite ve diğer hareket bozukluklarının yanı sıra Apraksi (motor fonksiyonları yerine getirememe) ve ataksi (koordine olmayan hareketler) yaygındır.
- Hiperventilasyon, aerofaji** (hava yutma) ve apne (solunumun durduğu dönemler) dahil olmak üzere nefes alma düzensizlikleri.
- Genellikle PEG (perkütan endoskopik gastrostomi) tüpüyle besleme gibi tıbbi müdahaleler gerektiren kabızlık ve yutma güçlükleri gibi gastrointestinal sorunlar.
Hastalığın Seyri ve Aşamaları:
Rett sendromunun ilerlemesi, Hagberg ve Witt-Engerström’ün çalışmalarına dayanarak tipik olarak dört aşamaya ayrılır:
Aşama 1: Erken Başlangıç (Yavaşlama Aşaması, 6-18 ay):
- İlk gelişim normal görünür, ancak ince işaretler ortaya çıkmaya başlar. Bunlar arasında daha yavaş motor gelişim, çevreye ilgide azalma ve sosyal katılımda azalma yer alır. Çocuğun baş çevresi tipik büyüme modellerinin gerisinde kalmaya başlar (mikrosefaliye yol açar).
2. Evre: Hızlı Yıkıcı Evre (1-4 yaş):
- Bu, el kullanımı ve dil başta olmak üzere edinilmiş becerilerin kaybı ile belirgindir. El yıkama stereotipleri ve tekrarlayan hareketler, artan sosyal geri çekilme ve otistik benzeri davranışlar ile birlikte belirgin hale gelir. Çığlık atma** ve sinirlilik dönemleri ortaya çıkabilir.
3. Aşama: Plato (Kararlı Aşama, 2-10 yıl):
- Bu evrede gerileme yavaşlar ve bazı sosyal etkileşim ve iletişim becerileri hafifçe iyileşebilir. Bununla birlikte, epileptik nöbetler, apraksi ve motor defisitler (örneğin, dengesiz yürüyüş) genellikle kötüleşir veya daha belirgin hale gelir.
Evre 4: Geç Motor Bozulma (10 yıl sonra):
- Kaba motor beceriler bozulmaya devam ederek ciddi motor bozukluklara yol açar. Hastaların çoğu tekerlekli sandalyeye bağlı hale gelir. Hareket kabiliyetindeki azalmaya rağmen nöbetler azalabilir ve sosyal ve bilişsel beceriler stabilize olabilir, hatta iyileşebilir. Bununla birlikte, hareket daha katı hale gelir ve kas erimesi yaygındır.
Tedavi ve Yönetim:
Şu anda Rett sendromu için tedavi yok ve tedavi öncelikle semptomları yönetmeye ve yaşam kalitesini iyileştirmeye odaklanıyor. Temel müdahaleler şunları içerir:
Terapiler:
- Hareketliliği sürdürmek, kas gücünü artırmak ve amaca yönelik el kullanımını teşvik etmek için fizik tedavi ve meslek terapisi.
- İletişim becerilerini geliştirmek için konuşma terapisi, ancak birçok birey sözel bozukluklar nedeniyle alternatif iletişim yöntemlerine güvenecektir.
- Müzik terapisi** ve hipoterapi (atlarla terapi) ruh halini, motor koordinasyonu ve sosyal katılımı iyileştirmede faydalar göstermiştir.
Farmakolojik Müdahaleler:
- Antiepileptik ilaçlar (AED’ler)** genellikle nöbetleri yönetmek için kullanılır.
- apne** gibi solunum sorunları için sarizotan gibi ilaçlar klinik çalışmalarda araştırılmaktadır. Sarizotan, solunum anormalliklerini hafifletmek için tasarlanmıştır ve erken çalışmalarda umut vaat ettiğini göstermiştir.
Beslenme Desteği:
- Ciddi yutma güçlüklerinde, beslenme sağlamak ve aspirasyon pnömonisi riskini azaltmak için bir PEG tüpü kullanılabilir.
Spastisite ve Hareket Bozukluklarının Yönetimi:
- Fizyoterapi**, *plintleme* ve baklofen veya botulinum toksin enjeksiyonları gibi ilaçlar spastisite ve motor disfonksiyonun yönetilmesine yardımcı olabilir.
Gastrointestinal ve Solunum Sorunlarının İzlenmesi ve Tedavisi:
- Yutma bozuklukları**, *kabızlık* ve solunum anormallikleri gastrostomi ile beslenme ve solunum düzeninin izlenmesi gibi müdahalelerle sürekli yönetim gerektirir.
Gelecekteki Yönelimler:
Altta yatan MECP2 işlev bozukluğunu hedef alan gen tedavisi ve farmakolojik müdahaleler üzerine araştırmalar devam etmektedir. Deneysel tedaviler MECP2 işlevini geri kazandırmayı veya eksikliğinin aşağı yönlü etkilerini hafifletmeyi amaçlamakta ve gelecekte daha etkili tedaviler için umut vermektedir.
İleri Okuma
- Rett, A. (1966). “On a remarkable syndrome of cerebral atrophy associated with hyperammonemia in childhood.” Wiener Medizinische Wochenschrift, 116(37), 723-726.
- Hagberg, B., et al. (1983). “A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: report of 35 cases.” Annals of Neurology, 14(4), 471-479.
- Amir, R. E., et al. (1999). “Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2.” Nature Genetics, 23(2), 185-188.
- Chahrour, M., et al. (2008). “MeCP2, a key contributor to neurological disease, activates and represses transcription.” Science, 320(5880), 1224-1229.
- Neul, J. L., et al. (2010). “Rett syndrome: Revised diagnostic criteria and nomenclature.” Annals of Neurology, 68(6), 944-950.