1960’lar ve 1970’lerde, epilepsi uzmanlarının önünde hâlâ tam adını koyamadıkları bir hasta kümesi duruyordu: çocukluk ya da ergenlikte başlayan, giderek ağırlaşan miyokloniler, dirençli nöbetler, sarsak bir yürüyüş, kas güçsüzlüğü ve çoğu zaman açıklanamayan nörolojik çöküş. O dönem bu tabloyu, “progresif miyoklonik epilepsiler” adı altında toplu bir sepet teşhis olarak kullanıyorlardı; sepetin içinde Unverricht–Lundborg, Lafora, nöronal seroid-lipofuskinozlar ve o zamanlar henüz adı olmayan, fakat belirgin biçimde farklı duran bazı hastalar vardı.
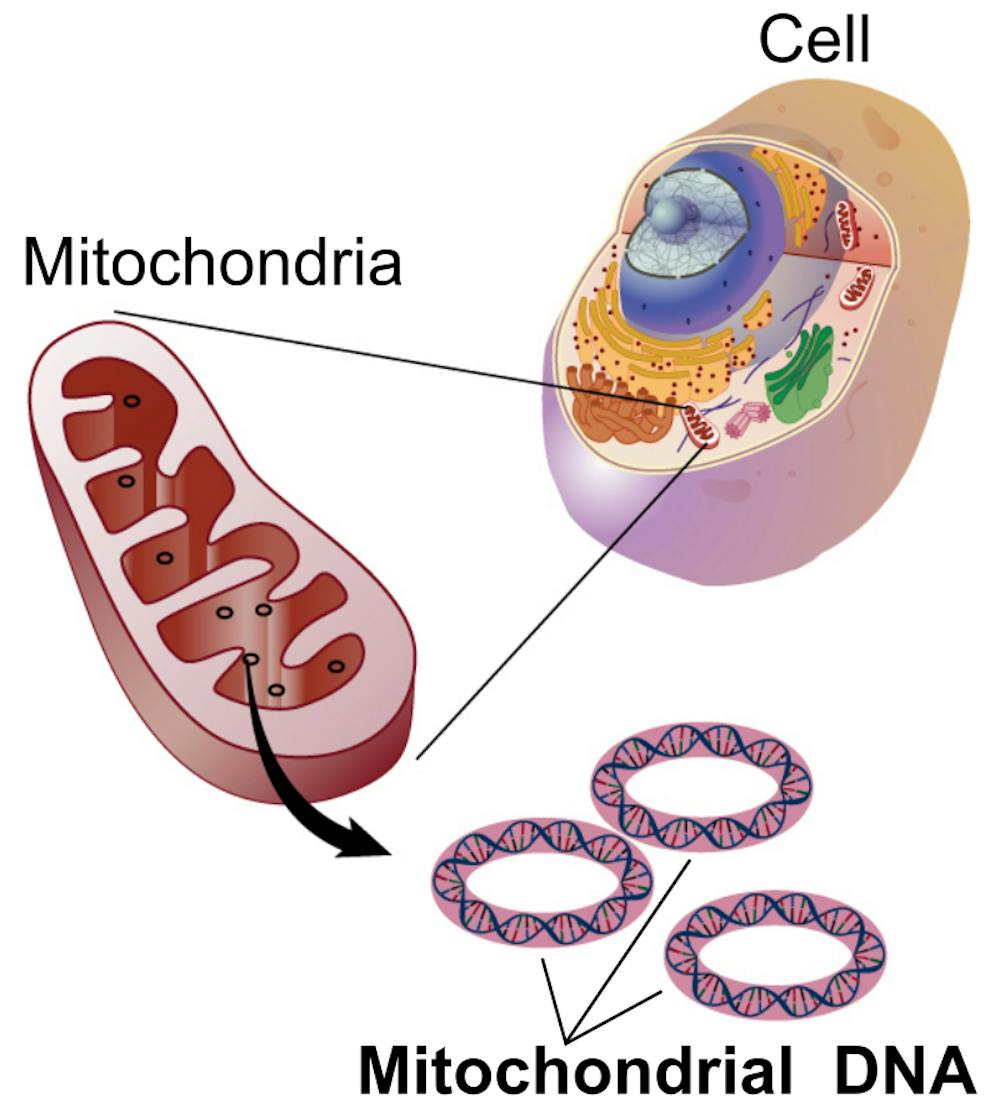
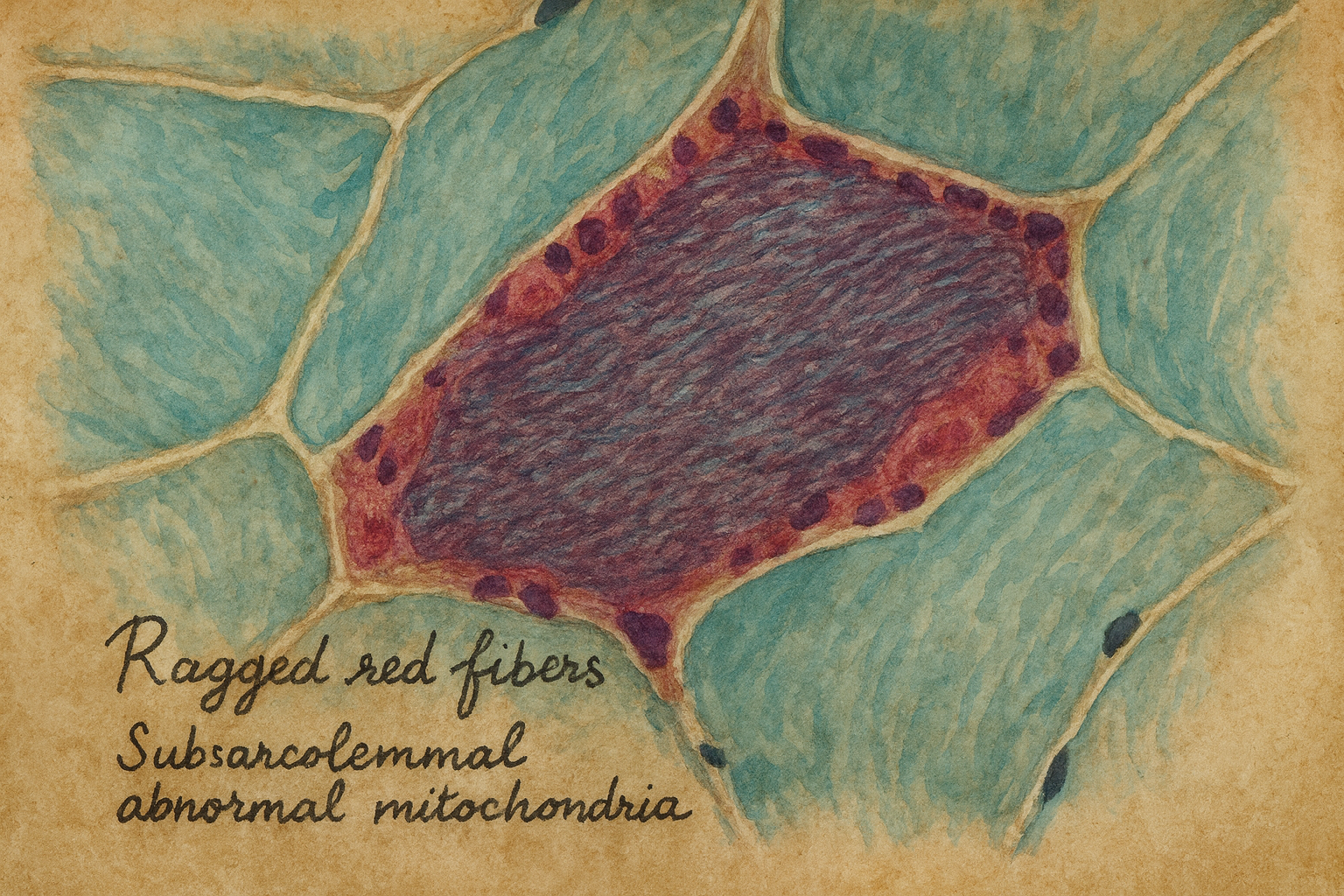
Nöropatologlar aynı yıllarda kas dokusunu yeni boyalarla inceleyerek ilginç bir ayrıntı fark ediyordu: kimi hastalarda bazı kas lifleri, modifiye Gomori trikrom boyasında alışılmadık derecede kaba, düzensiz ve kırmızı görünüyordu – subsarkolemmal alanda mitokondri yığılmasıyla oluşan bu görüntüye İngilizce “ragged red fibers”, yani “paçalı/düzensiz kırmızı lifler” adı verildi. Başlangıçta bu bulgu “mitokondriyal miyopati” denen geniş bir gruba işaret ediyor, fakat epilepsiyle arasındaki bağ henüz kurulamıyordu.
1980’lerin ikinci yarısına gelindiğinde, klinik nörologlar ile kas biyopsisi çalışan patomorfologlar daha yakından konuşmaya başladı. Avustralya’dan ve Avrupa’dan bildirilen olgu serilerinde, belirgin miyoklonik epilepsi tablosu olan bazı hastalarda kas biyopsisinde sistematik olarak ragged red fibers görülüyordu. Bu dikkatli gözlemlerin doruk noktası, 1989’da Berkovic ve çalışma arkadaşlarının Brain dergisinde yayımladıkları kapsamlı seri oldu: farklı yaşlarda başlamış, çoğu aynı aileye ait 13 hastayı ayrıntılı klinik, patolojik, biyokimyasal ve görüntüleme yöntemleriyle incelediler; tablo, miyoklonik epilepsi, ataksi, kas zayıflığı ve kas biyopsisinde ragged red fibers ile tutarlı, özgün bir sendrom olarak tarif edildi.
Bu makaleyle birlikte “myoclonus epilepsy with ragged-red fibres” ifadesi literatürde net bir sendrom adını almaya başladı. Ancak 1989’daki bu tanım hâlâ etiolojik olarak “agnostikti”: patolojik süreç mitokondriye işaret ediyor, fakat ortada ne spesifik bir gen, ne de tanımlı bir moleküler defekt vardı.
Kırılma noktası, 1990’da Shoffner ve arkadaşlarının çalışmasıyla geldi. Araştırma ekibi, Berkovic’in tanımladığı MERRF ailelerinden ve başka merkezlerden gelen benzer olguların kas ve kan örneklerini moleküler düzeyde incelemeye koyuldu. O yıllarda yeni yeni yaygınlaşan mitokondriyal DNA (mtDNA) analizi tekniklerini kullanarak, mtDNA üzerindeki tRNA genlerini sistematik biçimde taradılar ve sonunda kritik bir bulguya ulaştılar: insan mitokondriyal DNA’sının 8344 numaralı nükleotid pozisyonunda, tRNA-Lisin geninde özgül bir A→G geçiş mutasyonu, MERRF ailelerinde tekrar tekrar ortaya çıkıyor, sağlıklı kontrollerde ise görülmüyordu.
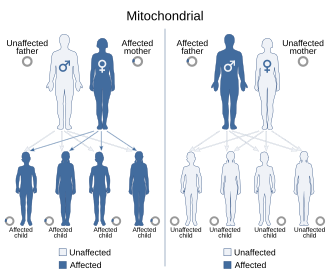
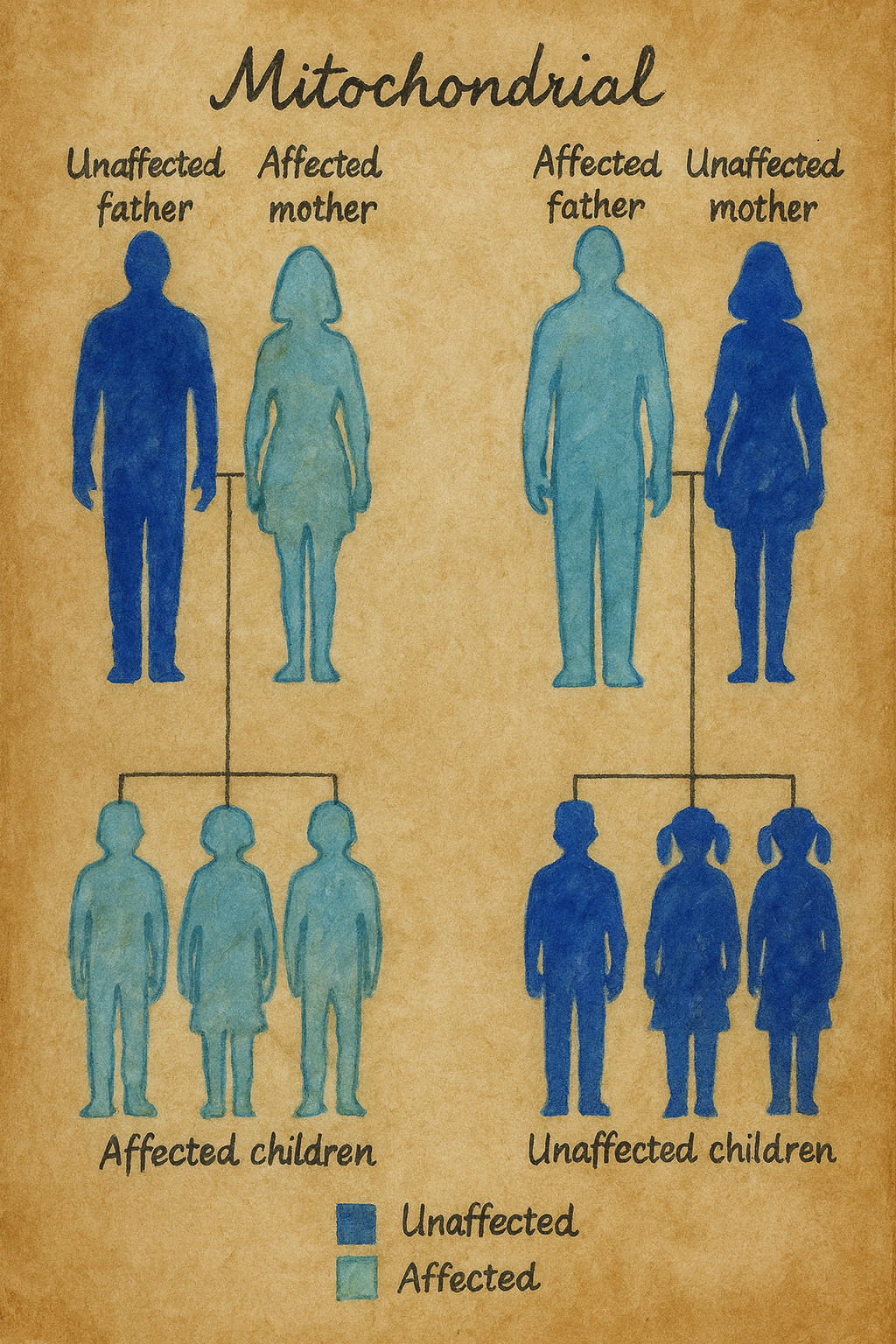
Bu keşif birden çok açıdan çarpıcıydı. Birincisi, MERRF ilk kez somut bir mtDNA nokta mutasyonuna bağlanmıştı; ikincisi, mutasyon tRNA genindeydi, yani disfonksiyon doğrudan mitokondriyal protein sentezinin “altyapısını” çarpıtıyordu; üçüncüsü, mutasyonun heteroplazmik olduğu, yani aynı bireyin hücrelerinde normal ve mutant mtDNA kopyalarının birlikte bulunduğu gösterilmişti. Ayrıca mutasyon, anneden aktarılan bir çizgi boyunca takip edilebiliyor, böylece klinikte zaten kuşkulanılan maternel kalıtım mekanik bir zemine oturuyordu.
Bu noktadan sonra 1990’ların başı MERRF için bir tür “haritalama çağına” dönüştü. Hammans ve diğer gruplar, farklı aileleri ve sporadik olguları inceleyerek 8344A>G mutasyonunun hangi heteroplazmi oranlarında hangi klinik fenotiplere yol açtığını sistematik biçimde analiz etmeye girişti. Bazı taşıyıcılarda ağır miyoklonik epilepsi ve erken ölüme giden diffüz ensefalomiyopati varken, aynı mutasyonu taşıyan başka akrabalarda klinik olarak sadece hafif kas zayıflığı veya simetrik lipomatoz görülmesi, genetikçiler ve nörologları hem büyüledi hem de zorladı. Bu çalışmalar, MERRF’nin sadece “bir sendrom” değil, mutant mtDNA oranına, doku dağılımına ve çevresel yüklenmelere göre şekillenen bir fenotipler spektrumu olduğunu gösterdi.
1990’ların ortalarından itibaren ilgi, “bu mutasyon var” demekten “bu mutasyon hücre içinde ne yapıyor?” sorusuna kaydı. Enriquez ve farklı gruplar, m.8344A>G mutasyonu taşıyan hücre dizilerini ve hasta kas dokularını karşılaştırmalı olarak incelediler. Mitokondrideki tRNA^Lys moleküllerinin aminoasilasyon kapasitesinin belirgin şekilde azaldığı, bunun da mitokondriyal protein sentezinde erken duraklamalara ve kısalmış polipeptid zincirlerine yol açtığı gösterildi. Özellikle kompleks I ve IV gibi solunum zinciri komplekslerinin alt birimlerinde bu üretim bozukluğu belirgindi; sonuç, düşmüş oksidatif fosforilasyon etkinliği, artmış oksijen radikali üretimi ve nöron ile kas liflerinde kronik enerji açığıydı.
Bu mekanistik çalışmalar MERRF’yi nörolojide bir “model hastalık” hâline getirdi. Anne Chomyn ve diğer araştırmacılar, aynı mutasyonun taşıyıcı hücrelerdeki heteroplazmi oranına göre mitokondriyal membran potansiyeli, ATP üretimi, ROS düzeyi ve apoptoz duyarlılığı gibi parametrelerde kademeli değişiklikler yarattığını göstererek, MERRF mutasyonunu insan mitokondri biyolojisini anlamak için adeta deneysel bir merceğe dönüştürdüler.
Bu arada klinik gözlemciler boş durmuyordu. 1990’lar ve 2000’ler boyunca, dünyanın dört bir yanından yeni vaka raporları ve aile serileri yayımlandı. Bazı olgularda MERRF tablosuna, özellikle boyun ve omuz çevresinde multipl simetrik lipomlar eşlik ediyordu; bazı ailelerde ise ağır kardiyomiyopati ve aritmi, tabloyu nörolojik belirtilerden önce gölgede bırakıyordu.
Daha da ilginci, bazı hastalarda klasik MERRF triadıyla birlikte MELAS’a özgü stroke-benzeri epizodlar ve kortikal laktoasidoz atakları ortaya çıkıyordu. Bu olgular “MERRF/MELAS overlap sendromu” olarak adlandırıldı ve araştırmacıları şu soruyla yüzleştirdi: Aynı mitokondriyal mutasyon, neden birinde epilepsi–ataksi ağırlıklı MERRF, diğerinde ise stroke-benzeri olaylarla giden MELAS fenotipi yaratıyor? Yanıt yine heteroplazmi, dokuya özgü eşik değerler ve muhtemelen ikinci “modifiye edici” faktörlerin karmaşık bileşimine bağlandı.
2000’lere gelindiğinde MERRF artık klinik açıdan tanımlı, genetik etiolojisi bilinen ve mekanistik düzeyde hatırı sayılır ölçüde çözülmüş bir mitokondriyal ensefalomiyopatiydi. Ancak pratikte hastalar hâlâ çok temel sorularla karşı karşıyaydı: “Nöbetlerimi nasıl kontrol ederiz?”, “Kas zayıflığım için ne yapılabilir?”, “Çocuk sahibi olursam ne olur?”
Klinisyenler, mitokondriye görece daha “nazik” antiepileptik ilaçları sistematik olarak denemeye başladılar. Giderek artan klinik deneyim, miyoklonik nöbetlerin özellikle levetirasetam ve benzodiazepinler (özellikle klonazepam) ile nispeten daha iyi kontrol edilebildiğini, bazı hastalarda zonisamid ve topiramat gibi ilaçların da fayda sağladığını gösterdi. Buna karşılık valproat gibi klasik ilaçların mitokondriyal yağ asidi metabolizmasını bozarak karaciğer yetmezliği ve metabolik dekompansasyon riskini arttırdığı, bu nedenle MERRF ve genelde mitokondriyal epilepsilerde mümkün olduğunca kaçınılması gerektiği vurgulanmaya başlandı.
2010’lar ve 2020’ler boyunca yayımlanan derleme ve kılavuz niteliğindeki yazılar, MERRF’de epilepsi tedavisi için bir “önerilen hiyerarşi” oluşturdu: birinci basamakta levetirasetam ve benzodiazepinler, ikinci basamakta zonisamid/topiramat, her zaman mitokondri toksisitesi bilinen moleküllerden kaçınma ilkesiyle birlikte. Özellikle Finsterer’in 2017 ve devam eden yıllardaki derlemeleri, MERRF’li hastalarda antiepileptik ilaç seçiminin inceliklerini ayrıntılı biçimde özetleyerek klinik pratiği önemli ölçüde etkiledi.
Aynı dönemde mitokondriyal “destek” tedavileri de giderek standart bir arka plan uygulaması hâline geldi. Koenzim Q10, L-karnitin, riboflavin, tiamin ve çeşitli antioksidan kombinasyonlarının kas yorgunluğu, egzersiz intoleransı ve genel performans üzerinde mütevazı fakat bazen anlamlı klinik iyileşmeler sağlayabildiğine dair olgu serileri ve küçük çalışmalar yayımlandı; kanıt düzeyi sınırlı kalsa da, bu ajanlar düşük yan etki profilleri nedeniyle pratikte çoğu merkezde tedavi kombinasyonuna eklendi.
MERRF araştırmalarının bir başka kolu, kalp tutulumu ve anestezi yönetimi üzerine odaklandı. m.8344A>G mutasyonunu taşıyan hastalarda sessiz kardiyomiyopati ve ritim bozukluklarının sanılandan daha sık olduğu, bu nedenle düzenli kardiyolojik taramanın kritik önemde olduğu ortaya kondu. Anestezi alan MERRF’li hastalarda, yüksek doz ve uzun süreli propofol infüzyonları gibi mitokondriyal solunum zinciri üzerinde ağır yük oluşturan rejimlerin riskleri ayrıntılı biçimde analiz edilmeye başlandı; 2020’ler boyunca yayımlanan anestezi derlemeleri, bu hastalarda “enerji ekonomisi” gözeten, kısa etkili ve mitokondriye daha az yük bindiren stratejilerin benimsenmesini önerdi.
Son yıllarda MERRF’in hikâyesi bir kez daha yön değiştirdi ve deneysel tedavi yaklaşımları sahneye çıktı. Mitokondri biyolojisinin ayrıntılı anlaşılması, birkaç yeni fikir doğurdu:
Birincisi, “daha fazla ve daha sağlıklı mitokondri” üretmek: PGC-1α gibi mitokondriyal biyogenez ustalarını hedefleyen yaklaşımlar ve nikotinik asit gibi NAD⁺ öncüleriyle mitokondri sayısını ve fonksiyonunu artırma denemeleri, m.8344A>G mutasyonu taşıyan hücrelerde in vitro olarak test edildi.
İkincisi, “bozulmuş mitokondrileri seçici olarak ortadan kaldırmak”: 2022 civarında yayımlanan çalışmalar, rapamisin gibi mTOR yolaklarını modüle eden ilaçlarla mitofaji süreçlerini uyararak, yüksek heteroplazmili hücrelerde bile mitokondri fonksiyonunu kısmen iyileştirmenin mümkün olabileceğini gösterdi. Hem sitoplazmik fibroblastlarda hem de mitokondri DNA’sı taşımayan (ρ0) hücrelere mutant mtDNA eklenerek oluşturulan model sistemlerde, rapamisin tedavisinin oksidatif fosforilasyonu ve oksijen tüketim hızını belirli ölçüde düzeltebildiği bildirildi.
Bu in vitro başarılar, klinik uygulamaya hemen çevrilebilecek düzeyde olmasa da, MERRF için “tamamen çaresiz” algısını kıran ilk işaretlerden biri oldu. Paralel olarak, mitokondriyal gen tedavisi alanında, mutant mtDNA moleküllerini seçici olarak kesmeyi veya yok etmeyi hedefleyen nükleaz-temelli stratejiler (örn. mitokondriye yönlendirilmiş TALEN ve ZFN türevleri) üzerinde deneysel çalışmalar yürütülüyor; hedef, mutant moleküllerin oranını düşürerek heteroplazmi eşiğinin altına inmeyi sağlamak.
Bu bilimsel gelişmelerin üzerine, bir başka çizgi daha ekleniyor: üreme tıbbı ve mitokondriyel replasman teknikleri. Bazı ülkelerde, ağır mtDNA mutasyonu taşıyan kadınların çocuk sahibi olabilmesi için, çekirdek DNA’sını sağlıklı mtDNA taşıyan donör oosit sitoplazmasına taşıyan “üç ebeveynli IVF” yöntemleri etik tartışmalar eşliğinde uygulanmaya başlandı. MERRF mutasyonunu taşıyan aileler bu tartışmanın merkezinde yer alıyor; çünkü burada söz konusu olan, yalnızca bir bireyin tedavisi değil, aynı zamanda mutasyon yükünün gelecek kuşaklara aktarımını azaltma çabası.
Klinik tarafta ise öykü daha “gündelik” ama en az o kadar kritik sorular etrafında şekillenmeye devam ediyor. MERRF için hazırlanan son kılavuzlar ve derlemeler, nöbet yönetiminden rehabilitasyona, kardiyak izlemeden psikososyal desteğe kadar uzanan geniş bir bakım çerçevesi tanımlıyor. Son yıllarda yayımlanan derlemeler, MERRF’li hastalarda miyoklonik epilepsinin en iyi ihtimalle levetirasetam, benzodiazepinler ve bazı olgularda zonisamid ile kontrol altına alınabildiğini; antiepileptik ilaçların her değişiminde mitokondriyal toksisite riskinin ayrıca hesaba katılması gerektiğini vurguluyor.
Aynı dönemde, 2023’te dört kuşaklık bir ailede bildirilen ve neredeyse hiç merkezi sinir sistemi bulgusu göstermeyen, buna karşın m.8344A>G mutasyonunu taşıyan bireyler, araştırmacılara MERRF’in ne kadar esnek bir fenotip yelpazesine sahip olduğunu bir kez daha hatırlattı: aynı mutasyon, bir kuşakta ağır epilepsi ve erken ölümle, başka bir kuşakta ise görece minimal kas yakınmalarıyla sınırlı kalabiliyor.
Bugün, 2020’lerin ortasında MERRF’in hikâyesine baktığımızda, yaklaşık yarım yüzyıllık bir bilimsel “roman” görüyoruz:
Önce klinisyenlerin sezgileriyle başlayıp kas mikroskobisiyle desteklenen, adı bile olmayan bir sendrom; ardından 1989’da Berkovic’in çizdiği detaylı klinik portre; 1990’da Shoffner’ın mtDNA’daki 8344A>G mutasyonunu işaret etmesiyle gelen büyük açıklama; 1990’lar ve 2000’lerde heteroplazmi, genotip-fenotip ilişkileri ve mekanistik deneyler; 2010’larda mitokondri dostu epilepsi tedavi stratejilerinin şekillenmesi; 2020’lerde ise mitofaji, biyogenez, gen tedavisi ve üreme tıbbı ekseninde açılan yeni, daha cesur sorular.
MERRF hâlâ tedavi edilemeyen, seyri heterojen ve çoğu zaman ağır bir hastalık; fakat artık yalnızca karanlık, açıklanamayan bir progresif epilepsi sendromu değil. Bir yandan hastaların yaşam kalitesini iyileştirmeye dönük çok daha sofistike klinik stratejiler geliştirilmiş durumda; öte yandan mitokondri biyolojisinin derinliklerine inen deneysel çalışmalar, bu hikâyenin sonraki bölümlerinde “doğrudan hedefe yönelik” tedavilerin de sahneye çıkabileceğini düşündürüyor.









Yorum yazabilmek için oturum açmalısınız.