Konjenital Adrenal Hiperplazi (KAH)
I. Terminolojik Köken ve Kavramsal Çerçeve
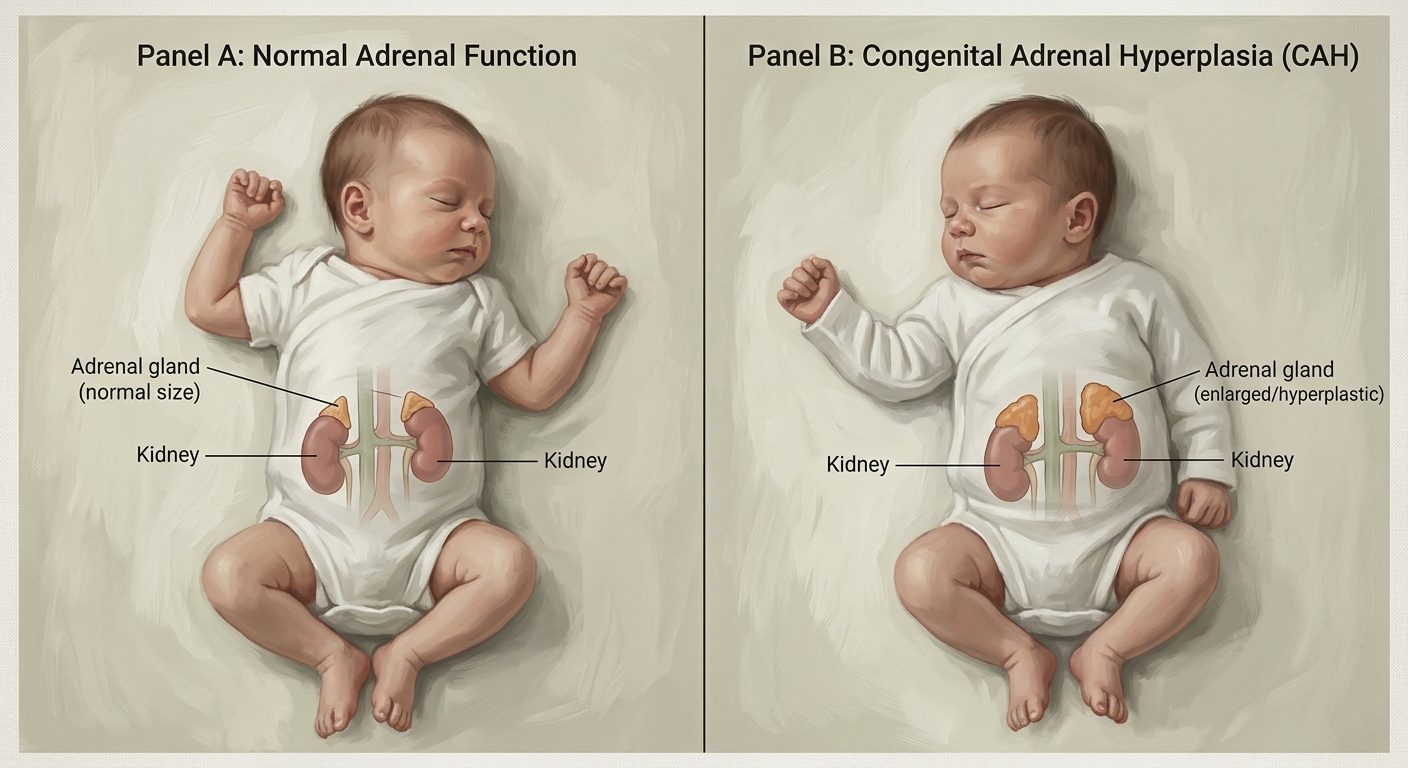
Konjenital Adrenal Hiperplazi (KAH) terimi, patolojik sürecin zamansal, anatomik ve morfolojik boyutlarını tanımlayan klasik tıp etimolojisinin bir ürünüdür. Latince kökenli congenitus (doğuştan gelen, birlikte doğan), hastalığın intrauterin yaşamda başlayan kalıtsal doğasını vurgular. Yine Latince ad (yakınında) ve renes (böbrekler) sözcüklerinin birleşiminden oluşan adrenal tanımı, etkilenen endokrin bezlerin böbrek üst kutbuyla olan topografik komşuluğuna işaret eder. Grekçe hyper (aşırı, üzerinde) ve plasis (oluşum, şekil verme) köklerinden türeyen hiperplazi ise, hücre sayısındaki artışa bağlı olarak bez volümünde meydana gelen karakteristik büyümeyi betimler. Bu etimolojik yapı, hastalığın temel patofizyolojik üçgenini özetler: Doğuştan gelen bir genetik defekt, böbreküstü bezlerinde trofik bir yanıta yol açar ve bez dokusunun anormal proliferasyonu ile sonuçlanır.
II. Evrimsel Biyoloji Perspektifinden Adrenal Yolağın Hassasiyeti
Adrenal korteks tarafından yürütülen steroidogenez faaliyeti, omurgalı canlıların sucul ekosistemlerden karasal habitata geçişinde kritik bir evrimsel adaptasyon olarak şekillenmiştir. Ozmotik dengenin sağlanmasında mineralokortikoidlerin (aldosteron), enerji metabolizmasının ve stres yanıtının düzenlenmesinde ise glukokortikoidlerin (kortizol) rolü, organizmanın homeostatik esnekliğini belirleyen temel unsurlardır.
KAH patogenezinde merkezi rol oynayan sitokrom P450 enzim sistemi (özellikle 21-hidroksilazı kodlayan CYP21A2 geni), memeli genomunun en polimorfik ve rekombinasyona en yatkın bölgelerinden biri olan majör histokompatibilite kompleksi (MHC) Sınıf III bölgesinde lokalizedir. Bu genomik kırılganlık, CYP21A2 geni ile onun yüksek oranda homolog fakat işlevsiz bir psödogeni (CYP21A1P) arasında meydana gelen gen dönüşümü ve eşit olmayan krossing-over olaylarına zemin hazırlar. Evrimsel süreçte, bu genetik varyantların heterozigot taşıyıcılarına, özellikle proinflamatuar sitokin yanıtlarının modülasyonu yoluyla bazı enfeksiyöz hastalıklara karşı seçici bir avantaj sağladığı hipotezi öne sürülmektedir. Ancak biallelik mutasyon varlığında ortaya çıkan şiddetli kortizol eksikliği ve kontrolsüz androjen fazlalığı tablosu, bu karmaşık genomik bölgenin evrimsel bedelini gözler önüne serer.
III. Güncel Bilimsel Anlayış: Moleküler Patofizyoloji ve Genetik Mimari
3.1. Enzimatik Defekt ve Hipotalamo-Hipofizer-Adrenal Aksın Disregülasyonu
Güncel tıp literatüründe KAH, adrenal kortekste kolesterolden kortizol biyosentezini katalize eden bir dizi enzimden herhangi birinin kalıtsal eksikliği olarak tanımlanır. Vakaların yaklaşık %95’inden sorumlu olan 21-hidroksilaz eksikliği, temel biyokimyasal akışta bir darboğaz oluşturur. Bu enzimatik blokaj, 17-hidroksiprogesteronun 11-deoksikortizole dönüşümünü engelleyerek serum kortizol seviyelerinin kronik olarak düşük seyretmesine neden olur. Kortizol tarafından sağlanan negatif geri bildirimin ortadan kalkması, hipofiz ön lobundan adrenokortikotropik hormon (ACTH) salınımında kontrolsüz bir artışa yol açar.
3.2. Hiperplazi ve Androjen Sentezine Şant Mekanizması
Yüksek ACTH seviyeleri, adrenal korteks hücreleri üzerinde sürekli bir trofik uyarı oluşturarak hiperplazi olarak adlandırılan doku büyümesine neden olur. Bununla eş zamanlı olarak, ACTH uyarısı, enzimatik blokajın proksimalinde biriken steroid prekürsörlerinin (başlıca 17-hidroksiprogesteron ve progesteron) alternatif metabolik yollara yönlendirilmesine sebep olur. Bu prekürsörler, 21-hidroksilasyon gerektirmeyen yolaklar üzerinden androstenedion ve testosteron gibi potent androjenlere dönüştürülür. Bu patolojik şant mekanizması, hastalığın klinik belirtilerinin büyük kısmından sorumlu olan intrauterin ve postnatal androjen fazlalığı durumunu yaratır.
IV. Klinik Fenotipler ve Tanısal Algoritmalar
KAH klinik prezentasyonu, altta yatan enzim defektinin rezidüel aktivitesine bağlı olarak geniş bir fenotipik yelpaze sergiler.
4.1. Klasik Form: Tuz Kaybettiren ve Basit Virilizan Varyantlar
Bu form, enzim aktivitesinin ciddi düzeyde düşük olduğu veya hiç olmadığı durumları kapsar.
- Tuz Kaybettiren Varyant: Aldosteron sentezindeki derin eksiklik sonucu yenidoğan döneminde ortaya çıkar. Hiponatremi, hiperkalemi, metabolik asidoz ve hipovolemik şok ile karakterize adrenal kriz tablosu, acil müdahale edilmediği takdirde hayatı tehdit eden bir durumdur. Bebeklerde emmede zayıflık, kusma, letarji ve tartı alamama gibi nonspesifik bulgularla prezente olur.
- Basit Virilizan Varyant: Mineralokortikoid aktivitesi görece korunmuştur ancak intrauterin androjen maruziyeti belirgindir. Dişi fetüslerde (46,XX), dış genital yapının değişen derecelerde maskülinizasyonu gözlenir; labioskrotal füzyon, klitoromegali ve ürogenital sinüs oluşumu tipiktir. İç genital organlar (uterus, overler, fallop tüpleri) Müllerian kanaldan normal gelişimini sürdürdüğü için, bu bireyler potansiyel fertiliteye sahiptir. Erkek bebeklerde ise dış genital yapı doğumda normaldir; ancak aşırı androjenler ilerleyen aylarda psödopüberte prekoks bulgularına (penil büyüme, pubarş, kemik yaşında dramatik ilerleme) yol açar.
4.2. Klasik Olmayan veya Geç Başlangıçlı Form
Daha hafif bir enzim eksikliği ile karakterize olan bu form, çocukluk çağının ilerleyen dönemlerinde, ergenlikte veya yetişkinlikte androjen fazlalığının kutanöz ve reprodüktif belirtileri ile ortaya çıkar. Klasik olmayan KAH, hirsutizm, tedaviye dirençli akne vulgaris, menstrual düzensizlikler (oligomenore veya amenore), anovulatuar infertilite ve erken pubarşın önemli ve sıklıkla atlanan bir nedenidir.
4.3. Tanısal Yaklaşım Prensipleri
- Yenidoğan Taraması: Gelişmiş ülkelerde rutin olarak uygulanan yenidoğan topuk kanı taramasında 17-hidroksiprogesteron düzeyinin ölçülmesi, tuz kaybettiren formun presemptomatik tanınmasını sağlayarak adrenal krize bağlı mortaliteyi önlemede kritik rol oynar.
- ACTH Stimülasyon Testi: KAH tanısında altın standart yöntemdir. Sentetik ACTH (kortrosin) enjeksiyonu öncesi ve sonrası alınan serum örneklerinde 17-hidroksiprogesteron ve diğer prekürsör steroidlerin düzeylerindeki abartılı yükselme, tanıyı kesinleştirir ve hastalığın tipini belirler.
- Moleküler Genetik Analiz: CYP21A2 geninin dizi analizi, spesifik mutasyonların tanımlanmasını, genotip-fenotip korelasyonunun yapılmasını ve aileye doğru genetik danışmanlık verilmesini mümkün kılar.
V. Klinik Yönetim ve Terapötik Stratejiler
KAH tedavisi, eksik olanın yerine konması ve fazla olanın baskılanması şeklinde özetlenebilecek ikili bir strateji üzerine kuruludur. Tedavi, multidisipliner bir yaklaşım gerektirir.
5.1. Glukokortikoid Replasmanı
Tedavinin temel taşı, eksik kortizolün yerine konmasıdır. Pediatrik popülasyonda büyüme üzerindeki olumsuz etkileri daha az olduğu için genellikle kısa etkili hidrokortizon tercih edilir. Erişkinlerde ise daha uzun etkili preparatlar (prednizolon veya deksametazon) kullanılabilir. Tedavinin doz ayarı son derece hassas bir dengedir; yetersiz tedavi androjen fazlalığına ve virilizasyona, aşırı tedavi ise iyatrojenik Cushing sendromu bulgularına (obezite, osteopeni, büyüme geriliği) neden olur. İdeal doz, serum 17-hidroksiprogesteron ve androstenedion düzeylerini normalin hafif üst sınırında tutan en düşük dozdur.
5.2. Mineralokortikoid Replasmanı
Tuz kaybettiren formda, aldosteron eksikliğini kompanse etmek için tedaviye sentetik mineralokortikoid olan fludrokortizon eklenir. Bu tedavi, elektrolit dengesini, plazma renin aktivitesini ve kan basıncını normal sınırlarda tutmayı amaçlar. Bebeklik döneminde sıklıkla oral sodyum klorür desteğine de ihtiyaç duyulur.
5.3. Cerrahi Müdahale ve Psikososyal Destek
Ağır genital maskülinizasyonu olan 46,XX KAH olgularında, multidisipliner bir ekip (pediatrik endokrinolog, çocuk cerrahı, çocuk psikiyatristi, tıbbi etik uzmanı) tarafından değerlendirilerek feminizan genitoplasti ameliyatı planlanabilir. Cerrahinin zamanlaması ve kapsamı, ailenin tam bilgilendirilmiş onamı ile bireyselleştirilmiş bir karar sürecini gerektirir. Ayrıca, androjen maruziyetinin prenatal nörogelişim üzerindeki potansiyel etkileri ve kronik hastalık yönetiminin getirdiği zorluklar nedeniyle, bireylere ve ailelerine yaşam boyu sürecek psikososyal destek sağlanması esastır.
5.4. Fertilite ve Gebelik Yönetimi
İyi kontrollü KAH’lı kadınlarda, optimum glukokortikoid tedavisi ile ovulatuar siklusların sağlanması ve spontan gebelik elde edilmesi mümkündür. Gebelik sırasında glukokortikoid dozunun dikkatli bir şekilde ayarlanması, fetal virilizasyon riskini minimize etmek için kritik öneme sahiptir. Gebelik planlayan çiftlere, taşıyıcılık taraması ve prenatal tanı seçenekleri hakkında kapsamlı genetik danışmanlık verilmelidir.
Keşif
Tıp tarihinin en büyüleyici keşif yolculuklarından biri, insan vücudunun en sessiz fakat en hayati bezlerinden ikisinin, böbreklerin hemen üzerinde adeta birer nöbetçi gibi konumlanmış adrenal bezlerin gizemini çözmeye yönelik entelektüel çabayla başlar. Bu yolculuk, Rönesans’ın anatomi salonlarından modern moleküler biyolojinin ışıltılı laboratuvarlarına uzanan, merak, tesadüf ve kararlılıkla örülmüş bir insanlık destanıdır. Hikâyenin merkezinde yer alan Konjenital Adrenal Hiperplazi (KAH), başlangıçta anlaşılamayan, çelişkili bulgularla dolu bir klinik bilmece olarak hekimlerin karşısına çıkmıştır.
Hikâyemiz, on altıncı yüzyılda, Bartolomeo Eustachi’nin adrenal bezleri ilk kez tanımlayıp onlara “böbreküstü cisimcikleri” adını vermesiyle sessiz bir giriş yapar. Ancak bu küçük bezlerin işlevi, neredeyse üç yüz yıl boyunca karanlıkta kalmaya mahkûmdu. On dokuzuncu yüzyılın ortalarına gelindiğinde, Guy’s Hospital’dan Dr. Thomas Addison, adrenal bezlerin tüberküloz gibi hastalıklarla harap olduğu ve hastaların derin bir halsizlik, ciltte bronzlaşma ve nihayetinde ölümle sonuçlanan bir tablo sergilediği çarpıcı gözlemlerini yayımladı. Addison’ın 1855 tarihli monografisi, bu küçük bezlerin yaşam için mutlak surette gerekli olduğunu gösteren ilk güçlü kanıttı. Ardından, 1856’da Brown-Séquard’ın adrenalektomi yapılan hayvanların hızla öldüğünü göstermesi, bu bilgiyi deneysel olarak perçinledi. Ancak KAH’ın bir hastalık antitesi olarak belirmesi için, bu bezlerin sadece eksikliğinin değil, anormal işleyişinin de dramatik sonuçlar doğurabileceğinin anlaşılması gerekiyordu.
Bu anlayışa giden yol, yirminci yüzyılın başlarında, birbiriyle görünüşte bağlantısız iki klinik gözlemin bir araya gelmesiyle aydınlanmaya başladı. Bir yanda, hekimler “psödohermafroditizm” olarak adlandırılan, dış genital yapıları iç üreme organlarıyla uyumsuz görünen bebeklerle karşılaşıyordu. Özellikle, iç anatomisi dişi olan bir bebeğin dış genitalyasının belirgin şekilde erkeksi özellikler göstermesi, tıbbi bir muamma olarak duruyordu. Öte yandan, başka bir grup çocuk, henüz bebeklik veya erken çocukluk döneminde şiddetli kusma, ishal, bitkinlik ve hızlı bir çöküşle seyreden ve çoğunlukla ölümle sonuçlanan gizemli bir “tuz kaybı” sendromuyla kaybediliyordu. Bu iki ayrı uçurumun aynı dağın iki yamacı olduğunu ilk sezen ve bu bağlantıyı bir hipotez haline getiren kişi, İtalyan patolog Luigi De Crecchio oldu. De Crecchio, 1865 yılında, dış genitalyası belirgin şekilde erkeksi olmasına rağmen otopsisinde uterus, overler ve fallop tüpleri bulunan bir bireyin detaylı anatomik incelemesini yayımladı. Bu, bugün geriye dönüp baktığımızda, bir 21-hidroksilaz eksikliği vakasının literatürdeki ilk ayrıntılı tasvirlerinden biri olarak kabul edilir. De Crecchio’nun bu gözlemi, tohumun toprağa düştüğü andı.
Ancak bu tohumun filizlenip bir bilgi ağacına dönüşmesi için bir yüzyıl daha beklemek gerekecekti. Yirminci yüzyılın ilk yarısı, steroid biyokimyasının altın çağıydı. Araştırmacılar, adrenal korteksten izole edilen gizemli maddelerin peşindeydi. İsviçreli kimyager Tadeus Reichstein ve Amerikalı biyokimyacı Edward Calvin Kendall, neredeyse eş zamanlı olarak kortizonu saflaştırmayı başardılar ve bu keşifleri onlara 1950 Nobel Fizyoloji veya Tıp Ödülü’nü kazandırdı. Kortizonun romatoid artrit gibi enflamatuar hastalıklardaki dramatik etkisi tüm dünyada yankı uyandırırken, pediatrik endokrinolojinin öncüleri bu yeni hormonun ışığında eski klinik bilmeceleri yeniden incelemeye başladılar. Bu noktada, sahneye iki dev isim çıkar: Baltimore’daki Johns Hopkins Hastanesi’nden Dr. Lawson Wilkins ve New York’taki Cornell Üniversitesi Tıp Fakültesi’nden Dr. Alfred Bongiovanni.
Wilkins, pediatrik endokrinolojinin kurucu babası olarak anılır. Onun dikkatli klinik gözlemleri, konjenital adrenal hiperplazili çocukların idrarında anormal miktarlarda androjenik steroid (17-ketosteroidler) bulunduğunu gösterdi. Bu, aşırı erkekleşmenin biyokimyasal kanıtıydı. Ancak asıl zekice sıçrama, bu çocuklara yeni keşfedilen “mucize ilaç” kortizonu vermeyi denemesiyle gerçekleşti. Wilkins ve meslektaşları, 1950 yılında yayımladıkları ufuk açıcı bir makalede, KAH’lı bir çocuğa kortizon verildiğinde idrardaki 17-ketosteroid seviyelerinin dramatik bir şekilde düştüğünü bildirdiler. Bu gözlem, hastalığın patofizyolojisini aydınlatan bir şimşek çakmasıydı. Wilkins, adrenal bezlerin birincil sorununun kortizol üretememek olduğunu, bunun da hipofiz bezini aşırı miktarda ACTH salgılaması için uyardığını ve bu uyarının adrenal bezleri büyütüp (hiperplazi) mevcut enzim yollarını zorlayarak aşırı androjen üretimine yol açtığını öne sürdü. Kortizon verilmesi, hipofizdeki ACTH üretimini baskılayarak bu kısır döngüyü kırıyordu. Bu, endokrinolojide negatif geri bildirim prensibinin klinik bir hastalığa uygulanan ilk ve en zarif örneklerinden biriydi. Bu keşif, hastalığın tedavisinin de temelini attı: Eksik hormonu yerine koymak, aşırı uyarıyı ve onun istenmeyen sonuçlarını ortadan kaldırıyordu.
Bu teorik çerçevenin kurulmasıyla birlikte, bilimsel merak bir sonraki katmana, moleküler düzleme yöneldi. Acaba hangi enzim eksikti? Bu sorunun cevabı, biyokimyasal yolakların haritalanmasıyla geldi. Araştırmacılar, KAH’lı hastaların adrenal dokularında ve idrarlarında, kortizol sentezi için gerekli bir ara basamak olan 17-hidroksiprogesteronun anormal derecede yüksek olduğunu tespit ettiler. Bu, tıkanıklığın tam olarak bu maddeyi bir sonraki aşamaya taşıyan 21-hidroksilaz enziminde olduğunu gösteriyordu. Artık hastalığın en yaygın formunun adı konmuştu: 21-Hidroksilaz Eksikliği. Bununla da kalınmadı; araştırmacılar, bazı bebeklerde görülen ölümcül tuz kaybının, aynı enzimin bir başka hayati işlevi olan aldosteron sentezindeki eksiklikten kaynaklandığını göstererek “tuz kaybettiren” ve “basit virilizan” formlar arasındaki klinik ayrımı biyokimyasal bir temele oturttular.
Hikâyenin genetik boyutunun aydınlanması ise 1970’lerin sonlarına doğru gerçekleşen bir başka entelektüel zaferdir. Araştırmacılar, KAH’ın ailelerde nasıl kalıtıldığını anlamaya çalışırken, beklenmedik bir keşif yaptılar: Hastalık, doku reddinde rol oynayan insan lökosit antijenleri (HLA) ile sıkı bir genetik bağlantı gösteriyordu. Bu, sorumlu genin, altıncı kromozomun kısa kolu üzerinde, HLA gen kompleksinin tam kalbinde yer aldığını işaret eden bir yol göstericiydi. Nihayet 1980’lerin ortalarında, moleküler genetik tekniklerinin gelişmesiyle birlikte, Perrin C. White ve meslektaşları, 21-hidroksilaz enzimini kodlayan CYP21A2 genini başarıyla klonladılar ve dizilimini belirlediler. Bu keşif, hastalığın tüm gizemini çözen anahtardı. Araştırmacılar, aktif genin hemen yanı başında, evrim sürecinde işlevini yitirmiş fakat dizi benzerliğini büyük ölçüde korumuş bir psödogenin (CYP21A1P) varlığını ortaya çıkardılar. İşte bu ikiz benzeri yapı, genomun en büyük kırılganlık noktalarından birini oluşturuyordu. Mayoz bölünme sırasında, aktif gen ile psödogen arasında meydana gelen hatalı gen dönüşümleri ve rekombinasyon olayları, CYP21A2 genine zararlı mutasyonların sızmasına neden oluyordu. Bu keşif, KAH’ın neden bu kadar yaygın bir kalıtsal hastalık olduğunu açıklamakla kalmadı, aynı zamanda genetik danışmanlık ve prenatal tanı için kesin bir moleküler temel sağladı.
Bu moleküler anlayış, klinik uygulamada devrim niteliğinde bir başka gelişmeyle el ele yürüdü: Yenidoğan Taraması. 1977 yılında, Hawaii’den Dr. Richard J. Schanler ve Dr. Kenneth L. F. Pueschel’in de aralarında bulunduğu bir grup araştırmacı, yenidoğanların topuklarından alınan kurutulmuş kan lekelerinde 17-hidroksiprogesteron düzeyinin ölçülebileceğini gösterdi. Bu basit, ucuz ve hızlı test, tuz kaybettiren KAH’lı bebekleri, hayatı tehdit eden bir adrenal krizle hastanelere başvurmadan günler veya haftalar önce, henüz tamamen sağlıklı görünürlerken tespit etme olanağı sundu. Bu, koruyucu hekimliğin en büyük başarı öykülerinden biridir. Bugün dünyanın dört bir yanındaki yenidoğan tarama programları, her yıl binlerce bebeğin hayatını kurtarmakta ve onları ağır zihinsel veya fiziksel engellerden korumaktadır.
Günümüzde KAH araştırmalarının sınırları, daha da incelikli sorulara yanıt aramaktadır. Bilim insanları, genotip ile fenotip arasındaki karmaşık ilişkiyi tam olarak çözmeye, aynı mutasyonu taşıyan bireylerin neden farklı klinik tablolar sergilediğini anlamaya çalışmaktadır. Araştırmalar, prenatal androjen maruziyetinin beyin gelişimi ve cinsiyet kimliği üzerindeki nöropsikolojik etkilerine odaklanmakta, bu hassas konuda bireylere ve ailelere daha iyi rehberlik edebilmeyi amaçlamaktadır. Ayrıca, glukokortikoid tedavisinin kemik sağlığı, kardiyovasküler riskler ve metabolik sendrom gibi uzun dönemli komplikasyonlarını en aza indirecek yeni tedavi rejimleri ve ilaç salınım sistemleri üzerinde yoğun çalışmalar sürdürülmektedir. Gen tedavisi ve adrenalde hedeflendirilmiş ilaç taşıyıcı sistemler gibi yenilikçi yaklaşımlar, gelecekte hastalığın nihai tedavisi için umut vaat etmektedir.
Luigi De Crecchio’nun otopsi masasındaki sessiz gözleminden, Wilkins’in kortizonla yaptığı cesur deneye ve oradan White’ın gen dizilimini çözmesine uzanan bu uzun ve dolambaçlı yol, bilimsel bilginin nasıl kümülatif bir şekilde ilerlediğinin, her bir keşfin bir sonraki için nasıl bir basamak oluşturduğunun ve insan merakının karanlığı nasıl adım adım aydınlattığının canlı bir kanıtıdır. Konjenital Adrenal Hiperplazi’nin keşif öyküsü, sadece bir hastalığın anlaşılması değil, aynı zamanda tıp biliminin kendi doğasına tutulmuş bir aynadır.
İleri Okuma
- Addison, T. (1855). On the Constitutional and Local Effects of Disease of the Supra-Renal Capsules. London: Samuel Highley.
- Brown-Séquard, C.-É. (1856). Recherches expérimentales sur les capsules surrénales. Comptes Rendus de la Société de Biologie, 8, 422–425.
- De Crecchio, L. (1865). Sopra un caso di apparenza virile in una donna. Il Morgagni, 7, 154–188.
- Kendall, E. C. (1949). The isolation and structure of adrenal cortical hormones. Journal of Biological Chemistry, 179(2), 1–13.
- Reichstein, T. (1950). Chemistry of the adrenal cortex hormones. Helvetica Chimica Acta, 33(6), 1467–1477.
- Wilkins, L., Lewis, R. A., Klein, R., & Gardner, L. I. (1950). Treatment of congenital adrenal hyperplasia with cortisone. Journal of Clinical Endocrinology & Metabolism, 10(4), 384–401. https://doi.org/10.1210/jcem-10-4-384
- Bongiovanni, A. M., & Root, A. W. (1963). The adrenogenital syndrome. New England Journal of Medicine, 268(25), 1391–1398. https://doi.org/10.1056/NEJM196306202682507
- White, P. C., New, M. I., & Dupont, B. (1987). Congenital adrenal hyperplasia. New England Journal of Medicine, 316(25), 1519–1524. https://doi.org/10.1056/NEJM198706183162507
- Speiser, P. W., & White, P. C. (2003). Congenital adrenal hyperplasia. New England Journal of Medicine, 349(8), 776–788. https://doi.org/10.1056/NEJMra021561
- Merke, D. P., & Bornstein, S. R. (2005). Congenital adrenal hyperplasia. The Lancet, 365(9477), 2125–2136. https://doi.org/10.1016/S0140-6736(05)66736-0
- White, P. C., & Speiser, P. W. (2000). Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocrine Reviews, 21(3), 245–291. https://doi.org/10.1210/edrv.21.3.0398
- New, M. I. (2006). Extensive clinical experience: nonclassical 21-hydroxylase deficiency. Journal of Clinical Endocrinology & Metabolism, 91(11), 4205–4214. https://doi.org/10.1210/jc.2006-1645
- Auchus, R. J. (2007). The genetics, pathophysiology, and management of human deficiencies of P450c21. Endocrinology and Metabolism Clinics of North America, 36(2), 259–276. https://doi.org/10.1016/j.ecl.2007.03.001
- Nimkarn, S., & New, M. I. (2010). Prenatal diagnosis and treatment of congenital adrenal hyperplasia. Hormone Research in Paediatrics, 74(6), 404–412. https://doi.org/10.1159/000313374
- Speiser, P. W., Arlt, W., Auchus, R. J., et al. (2018). Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: An Endocrine Society clinical practice guideline. Journal of Clinical Endocrinology & Metabolism, 103(11), 4043–4088. https://doi.org/10.1210/jc.2018-01865
- El-Maouche, D., Arlt, W., & Merke, D. P. (2017). Congenital adrenal hyperplasia. The Lancet, 390(10108), 2194–2210. https://doi.org/10.1016/S0140-6736(17)31431-9