

- Fent-: Bu önek, fentanil molekülünün bir parçası olan fenetil grubundan (C₆H₅-CH₂-CH₂-) türetilmiştir. Fenetil grubu, fentanilin kimyasal yapısının tanımlayıcı bir özelliği olan bir etil zincirine (-etil) bağlı bir benzen halkasından (fen-) oluşur.
- -An-: Merkezi segment “-an-“, bileşiğin amin grubuna atıfta bulunan kısaltılmış bir form veya organik bileşiklerin adlandırma kuralında basitçe bir bağlayıcı unsur olabilir.
- -Yl: Kimyasal isimlendirmede “-il” eki genellikle daha büyük bir molekülden türetilen bir radikal veya fonksiyonel grubu belirtir. Fentanil durumunda, bu, onun belirli işlevsel kısımlarını ve piperidin sınıfındaki diğer türevlerle ilişkisini temsil eder.
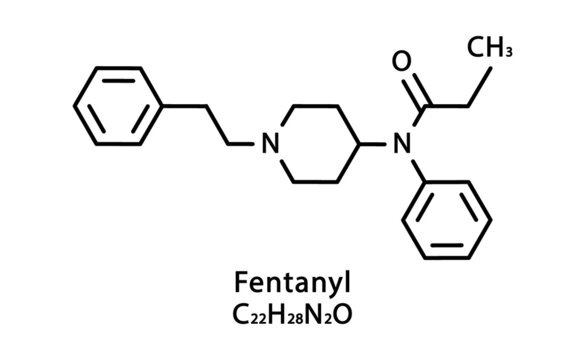
Bu nedenle, fentanil adı, sentetik opioid kimyası bağlamında işlevselleştirilmiş, fenetil grubu olan bir bileşik olarak kimyasal kimliğini yansıtır. Bu etimoloji, diğer sentetik opioidlerle yapısal ve işlevsel ilişkisini, özellikle petidin (meperidin)‘in bir analoğu olarak kökenini vurgular.
Fentanil, esas olarak şiddetli ağrının yönetimi için kullanılan yüksek etkili sentetik bir opioid analjeziktir. Pastiller, bukkal tabletler, dil altı tabletler, transdermal yamalar (örn. Durogesic®, jenerikler) ve enjekte edilebilir solüsyonlar dahil olmak üzere çeşitli formlarda ticari olarak mevcuttur. Bir narkotik olarak sınıflandırılması nedeniyle, fentanil sıkı reçete düzenlemelerine tabidir.

Moleküler Özellikler: Fentanil (C₂₂H₂₈N₂O, moleküler ağırlık = 336,5 g/mol) yüksek potansiyele sahip lipofilik, küçük bir moleküldür. Bu özellikler, deriden emilimini ve kan-beyin bariyerini geçerek MSS’ye ulaşma yeteneğini kolaylaştırır.
- Tuz formu, fentanil sitrat, suda çözünür ve belirli formülasyonlarda kullanılır.
- Metabolizma: Fentanil, karaciğer enzimi CYP3A4 tarafından metabolize edilir ve inaktive edilir, bu da onu önemli ilaç-ilaç etkileşimlerine duyarlı hale getirir.
Farmakolojik Mekanizma
Fentanilin analjezik etkileri, merkezi sinir sistemindeki (CNS) μ-opioid reseptörlerine seçici bağlanmasından kaynaklanır. Bu etkileşim ağrı sinyallemesini düzenler ve güçlü bir ağrı kesici üretir.
Uygulama
- Birincil Uygulama Yolu: Fentanil en yaygın olarak transdermal yamalar yoluyla uygulanır. Bu yöntem, uzun bir süre boyunca sürekli ilaç iletimi sağlar.
Endikasyonlar ve Kullanım
Fentanil şunlar için endikedir:
- Uzun süreli opioid tedavisi gerektiren hastalarda şiddetli kronik ağrının tedavisi.
- Opioid tedavisi alan ve bu tedaviye toleranslı olan kanser hastalarında ani ağrının yönetimi.
Yan Etkiler ve Riskler
Yaygın Yan Etkiler:
- Mide bulantısı
- Kusma
- Kabızlık
- Uyuşukluk
- Baş dönmesi
- Baş ağrısı
Ciddi Riskler:
- Solunum Depresyonu: Fentanil, özellikle aşırı doz durumlarında yaşamı tehdit eden solunum depresyonu riski taşır.
- Dozaj talimatlarına ve tıbbi yönergelere sıkı sıkıya bağlı kalmak, istenmeyen olay riskini en aza indirmek için kritik öneme sahiptir.
Yapı ve Kimyasal Özellikler
Fentanil, pethidine ile yapısal olarak ilişkilidir ve piperidin türevi bileşik sınıfına aittir. Temel formunda suda pratik olarak çözünmez ancak fentanil sitrat olarak tuz formunda suda çözünür hale gelir. Bu özellikler, dermal ve oral uygulama için çeşitli farmasötik formülasyonlarda çok yönlü kullanımına olanak tanır.
Önlemler ve İlaç Etkileşimleri
CYP3A4 aracılığıyla metabolizması göz önüne alındığında, fentanil aşağıdakilerle etkileşime girmeye eğilimlidir:
- CYP3A4 inhibitörleri (örneğin, belirli antifungal ilaçlar, makrolid antibiyotikler ve proteaz inhibitörleri), etkilerini ve toksisitesini artırabilir.
- Diğer MSS depresanları (örneğin, benzodiazepinler, alkol), sedasyonu ve solunum depresyonunu artırabilir.
Keşif

1950’ler–1960’lar: Keşif ve İlk Geliştirme
- 1959: Fentanil ilk olarak Dr. Paul Janssen ve Belçika’daki Janssen Pharmaceutica’daki ekibi tarafından sentezlendi. μ-opioid reseptörleri için yüksek potansiyele ve seçiciliğe sahip sentetik opioidler yaratma araştırma çabasının bir parçası olarak geliştirildi.
- 1960: Fentanil, özellikle cerrahi ortamlarda tıbbi kullanım için hızlı etkili bir anestezik olarak Sublimaze® ticari adı altında patentlendi.
1970’ler: Yaygın Klinik Kullanım
- 1972: Fentanil, Amerika Birleşik Devletleri’nde intravenöz anestezik olarak kullanım için onay aldı ve gücü ve hızlı başlangıcı nedeniyle anestezi uygulamalarında devrim yarattı.
- 1970’lerin sonu: Kullanımı, özellikle kanser hastalarında ve büyük ameliyatlar geçirenlerde şiddetli ağrı yönetimini de kapsayacak şekilde genişledi.
1980’ler: Yeni Dağıtım Sistemleri
- 1981: Kronik ağrısı olan hastalara sürekli ağrı kesici sağlamayı amaçlayan fentanil yamasının (transdermal sistem) geliştirilmesi başladı. Bu yenilik, sürekli ilaç dağıtımına izin vererek sık dozlama ihtiyacını azalttı.
1990’lar: Fentanil Yamasının Onayı
- 1990: FDA, ilk transdermal fentanil yamasını Durogesic® marka adı altında onayladı (daha sonra ABD’de Duragesic® olarak pazarlandı). Kronik ve kanserle ilişkili ağrının tedavisinde temel taş haline geldi.
2000’ler: Formülasyonların Genişlemesi
- 2001: Opioid toleranslı kanser hastalarında ani ağrıyı yönetmek için tasarlanmış oral fentanil pastillerinin (Actiq®) piyasaya sürülmesi. Bu, epizodik ağrı için daha hızlı etkili bir alternatif sağladı.
- 2006: Ani ağrı yönetimi için başka bir seçenek olan fentanil bukkal tabletlerin (Fentora®) onaylanması.
- 2009: Dil altı fentanil tabletlerinin ve diğer hızlı başlangıçlı formülasyonların onaylanması.
2010’lar: Artan Farkındalık ve Zorluklar
- 2010’lar: Fentanil, kötüye kullanım ve bağımlılık raporlarının artmasıyla opioid salgınında önemli bir oyuncu haline geldi. Yüksek potansiyeli (morfinden yaklaşık 50-100 kat daha güçlü) onu kötüye kullanım hedefi haline getirdi.
- 2013: Karfentanil gibi sentetik fentanil analogları yasadışı uyuşturucu pazarlarında görünmeye başladı ve opioid aşırı doz ölümlerinde keskin bir artışa katkıda bulundu.

2020’ler: Devam Eden Zorluklar ve Yenilikler
- 2020’ler: Kötüye kullanım risklerine rağmen fentanil, özellikle palyatif bakım ve kanser tedavisinde ağrı yönetimi için kritik bir ilaç olmaya devam ediyor.
- Devam Eden Araştırma: Analjezik etkinliği korurken daha güvenli dağıtım mekanizmaları ve kötüye kullanım potansiyeli azaltılmış analoglar geliştirme çabaları devam ediyor.
- Düzenleme ve Farkındalık: Dünya çapındaki hükümetler ve sağlık örgütleri, fentanilin kötüye kullanımını ve saptırılmasını azaltmak için daha sıkı düzenlemeler ve izleme programları uyguluyor.











Yorum yazabilmek için oturum açmalısınız.