Hematoloji, kan ve kan oluşturan dokuların incelenmesiyle ilgilenen tıp dalıdır. Kanla ilgili hastalıkların teşhisini, tedavisini ve önlenmesini içerir. Hematologlar bu alanda uzmanlaşmış tıp doktorlarıdır.
Hematolojinin temel çalışma alanlarından bazıları kan hücresi üretimi ve gelişimi, kan bozukluklarının nedenleri ve etkileri ve hastalıkları tedavi etmek ve önlemek için kan ürünlerinin kullanımıdır. Kanser tedavisi genellikle kan ve kan oluşturan dokuları etkilediğinden, hematologlar kanserli hastalarla da çalışabilir.
Hematolojide kullanılan tanı teknikleri arasında kan testleri, kemik iliği biyopsileri ve sitogenetik ve moleküler genetik testler yer alır. Kan bozuklukları için tedavi seçenekleri arasında ilaçlar, kan nakli ve bazı durumlarda kemik iliği veya kök hücre nakli yer alabilir. Hematologlar hastaneler, klinikler ve akademik araştırma kurumları da dahil olmak üzere çeşitli ortamlarda çalışabilirler.
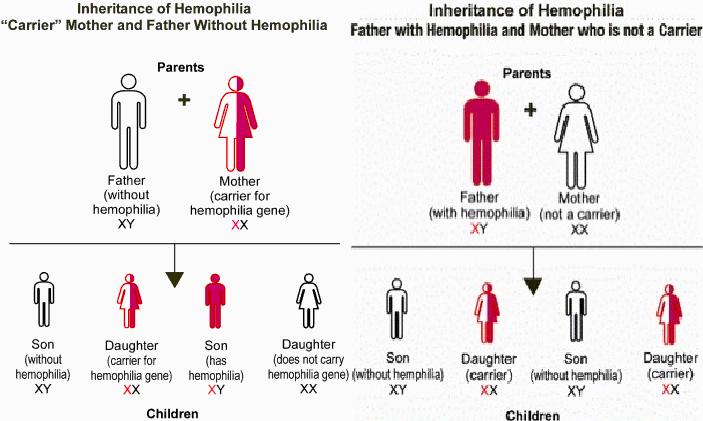
Sinonim: Haemophilia A, Hämophilie A, Bluterkrankheit A.
Bir kalıtsal kan hastalığıdır, kan pıhtılaşmasını hasarlı yapar. (bkz: Haem-o-philia)
Bu hastalık x kromozomal çekinik kalıtsal kan pıhtılaşmasını engeller.
Klasik hemofili bir kişide ağır ve uzamış kanama geliştiğinde gerçek anlamda etkin olan tek tedavi saflaştırılmış faktör VIII enjeksiyonudur.Faktör VIII çok pahalıdır ve yalnızca insan kanından çok az miktarlarda elde edilebildiğinden bulunması kısıtlıdır.
Tedavi stratejileri altta yatan nedene ve pansitopeninin ciddiyetine bağlıdır:
Destekleyici Tedavi
Eritrosit (EC) ve trombosit konsantrelerinin (TC) uygulanması.
Şiddetli nötropenide enfeksiyon profilaksisi (örn. antibiyotikler, antimikotikler, G-CSF).
Spesifik Tedavi
Genetik koşullar: Allojeneik hematopoetik kök hücre transplantasyonu.
İmmün ile ilişkili pansitopeni: İmmünosüpresif tedavi.
Etiyolojiye Özgü Müdahaleler
Beslenme eksikliklerinin giderilmesi (örn. B12 vitamini veya folik asit).
Enfeksiyon ve malignitelerin tedavisi.
Keşif
Üç ana kan hücresi tipinin (kırmızı kan hücreleri, beyaz kan hücreleri ve trombositler) tümünde ciddi bir azalma ile karakterize olan pansitopeni, zengin ve karmaşık bir gözlem ve keşif geçmişine sahiptir.
Erken Gözlemler
Eski Mısır (M.Ö. 1550): Bilinen en eski tıbbi metinlerden biri olan Papirüs Ebers, pansitopeniyi düşündüren yorgunluk, kanama ve enfeksiyonlar gibi semptomları tanımlamaktadır.
Ortaçağ Avrupası: “Kara Ölüm” (hıyarcıklı veba) 14. yüzyılda Yersinia pestis enfeksiyonunun yol açtığı kemik iliği hasarı nedeniyle yaygın pansitopeniye neden olmuştur.
19. Yüzyıl Gelişmeleri
Hematolojinin Doğuşu: Kan hücresi üretimi ve bozukluklarının anlaşılmasındaki ilerlemeler, pansitopeninin ayrı bir durum olarak tanınmasını sağlamıştır, ancak nedenleri büyük ölçüde bilinmemektedir.
20. Yüzyıl Buluşları
1900’lerin başları
“Pansitopeni” terimi ilk kez tıp literatüründe ortaya çıkmıştır.
1930‘lar
Kan hücresi üretim yeri olarak kemik iliğinin tanımlanması.
İlik fonksiyonunu etkileyen dış faktörlerin tanınması, pansitopeninin anlaşılmasına zemin hazırlar.
1940‘lar
Radyasyon ve bazı ilaçların pansitopeni nedeni olarak keşfedilmesi.
Bu bulgu, çevresel ve terapötik maruziyetleri kemik iliği yetmezliği ile ilişkilendirmiştir.
1950’ler-1960’lar
Aşağıdakiler de dahil olmak üzere teşhis araçlarının geliştirilmesi:
Doğrudan değerlendirme için Kemik iliği biyopsileri.
Pansitopeni ile ilişkili değişiklikleri tespit etmek için İleri kan testleri.
Geç 20. Yüzyıl Anlayışları
Fanconi Anemisinin Keşfi (1970’ler)
Nadir görülen bir genetik bozukluk olan Fanconi anemisi üzerinde çalışan araştırmacılar, hatalı DNA onarım mekanizmalarını tespit ederek pansitopenideki rolünü ortaya çıkardı.
Bu bulgu, radyasyon ve kimyasallar gibi kaynaklardan gelen DNA hasarının kan hücresi üretimini bozduğunu vurguladı.
Bu keşif, pansitopeni anlayışını genetik ve çevresel nedenleri de içerecek şekilde genişletmiştir.
Tanı ve Tedavide Devam Eden Gelişmeler
Yeni genetik test yöntemleri ve kemik iliği analiz teknikleri, pansitopeni etiyolojisine ilişkin daha derin bilgiler sağlamıştır.
Kök hücre nakilleri ve bağışıklık sistemini baskılayıcı tedaviler de dahil olmak üzere hedefe yönelik tedaviler hastalık sonuçlarını iyileştirmiştir.
Modern Anlayış
Pansitopeniye odaklanılması artık altta yatan spesifik nedenlerin tanımlanması etrafında dönmektedir, bunlar aşağıdakileri içerebilir:
Fanconi, G. (1927). Familial constitutional panmyelopathy (Fanconi anemia): Initial description and genetic implications.Zeitschrift für Kinderheilkunde, 44, 257–262.
Storb, R., & Thomas, E. D. (1983). Bone marrow transplantation: Historical developments and applications in bone marrow failure syndromes.Journal of the American Medical Association (JAMA), 249(17), 2294–2300. DOI: 10.1001/jama.1983.03330370056034
Bennett, J. M., & Catovsky, D. (1986). A historical perspective on aplastic anemia and related pancytopenias: Diagnosis and therapeutic approaches.American Journal of Hematology, 21(3), 269–278. DOI: 10.1002/ajh.2830210315
Alter, B. P., & Young, N. S. (1995). The bone marrow failure syndromes: Past, present, and future.Blood, 85(5), 1103–1110. DOI: 10.1182/blood.V85.5.1103.bloodjournal8551103
De Koning, H. W., & van Dongen, J. J. M. (1999). Advances in flow cytometry for the diagnosis of pancytopenia-related disorders.Cytometry Part B (Clinical Cytometry), 38(3), 150–159. DOI: 10.1002/(SICI)1097-0320(19990601)38:3<150::AID-CYTO4>3.0.CO;2-P
Young, N. S. (2000). Aplastic anemia and other bone marrow failure syndromes: From historical observations to molecular mechanisms.Blood, 96(2), 620–630. DOI: 10.1182/blood.V96.2.620
Savage, S. A., & Alter, B. P. (2009). Fanconi anemia and the discovery of DNA repair mechanisms.Nature Reviews Cancer, 9(10), 737–748. DOI: 10.1038/nrc2631
Ruggeri, A., & Gluckman, E. (2012). Historical advances in hematopoietic stem cell transplantation for bone marrow failure syndromes.Best Practice & Research Clinical Haematology, 25(2), 95–102. DOI: 10.1016/j.beha.2012.06.002
Myers, K. C., & Shimamura, A. (2015). Genetic advances in inherited bone marrow failure syndromes: Implications for pancytopenia diagnosis and treatment.British Journal of Haematology, 169(4), 373–387. DOI: 10.1111/bjh.13329
Kulasekararaj, A. G., & Marsh, J. C. W. (2016). Aplastic anemia and pancytopenia: Current concepts and controversies.Hematology/Oncology Clinics of North America, 30(3), 467–482. DOI: 10.1016/j.hoc.2016.01.005
Tolar, J., & Wagner, J. E. (2018). Gene therapy in Fanconi anemia and inherited pancytopenias: Historical perspective and future prospects.Blood, 131(3), 304–311. DOI: 10.1182/blood-2017-09-804930
Marsh, J. C. W., & Ball, S. E. (2019). Advances in the understanding and treatment of acquired pancytopenia syndromes.Haematologica, 104(5), 849–858. DOI: 10.3324/haematol.2019.215483
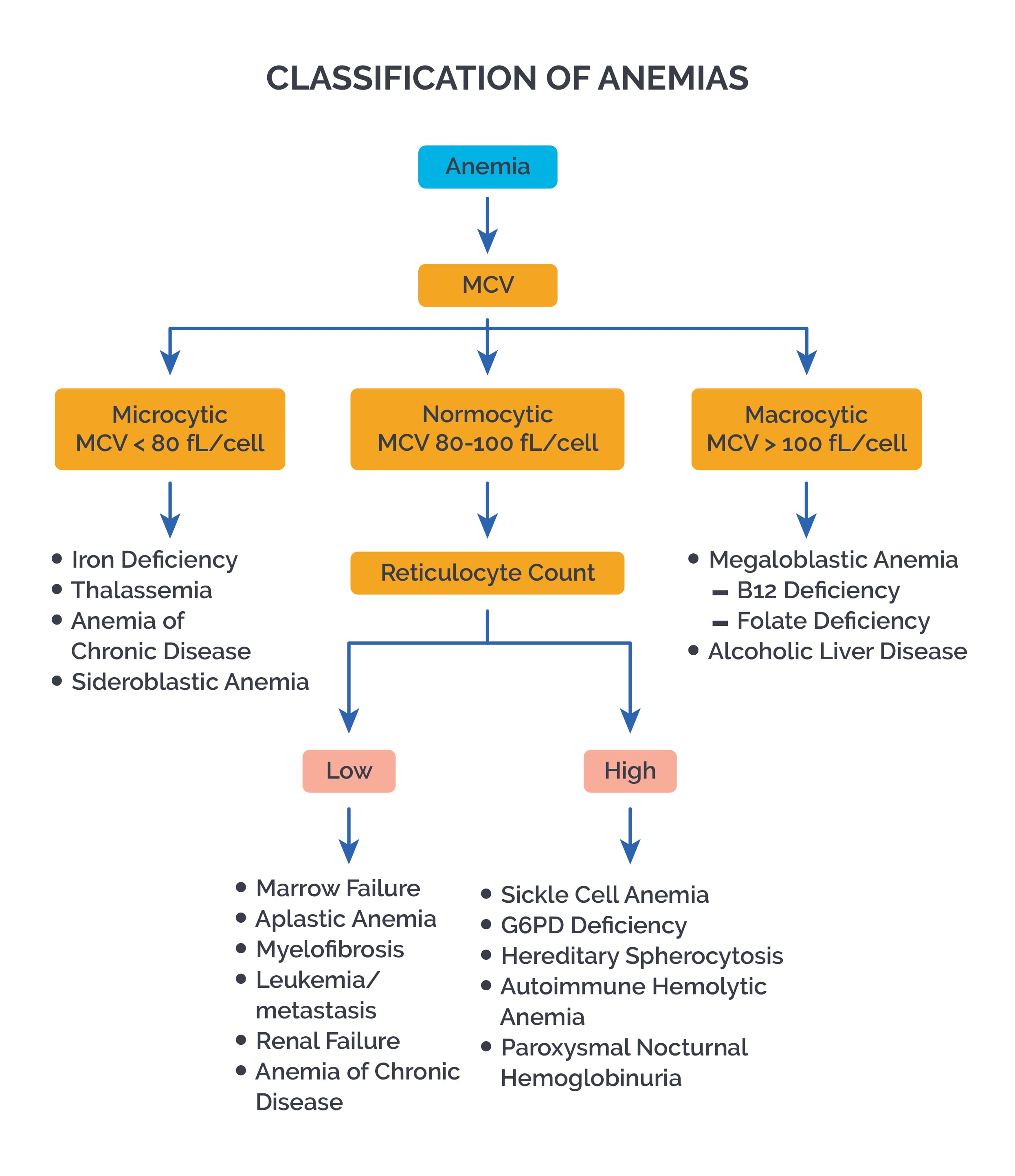
Makrositik anemi, ortalama eritrosit hacmi (MCV) 96 fl’den fazla olan anemidir.Makrositik anemi hemen hemen her zaman hiperkromiktir, bkz. hiperkromik makrositik anemi.
Sınıflandırma
Makrositik anemiler megaloblastik anemi ve megaloblastik olmayan anemi formlarına ayrılabilir.
Etiyoloji
Makrositer aneminin nedenleri çeşitlidir.
Megaloblastik anemi, bir DNA sentez bozukluğundan kaynaklanır. Bunun nedeni çoğunlukla:
B12 vitamini eksikliği (örn. pernisiyöz aneminin bir parçası olarak)
folik asit eksikliği
Megaloblastik olmayan anemi formları şu durumlarda bulunur:
miyelodisplastik sendrom
alkol kötüye kullanımı
miksödem
hemoliz
İlaç almak (Ara-C, hidroksiüre, antiviraller, antikonvülsanlar)
Semptom
Aneminin ciddiyetine bağlı olarak tipik klinik belirtiler şunlardır:
solgunluk
Yorgunluk, düşük performans, yorgunluk
baş ağrısı, baş dönmesi
takipne, nefes darlığı
taşikardi
Teşhis
Prensip olarak, makrositer anemi tanısı için MCV ( 96 fl) ve Hb değerinin (♂ 13,5 g/dl; ♀ 12,5 g/dl) belirlenmesi yeterlidir. Etiyolojiyi belirlemek ve diğer anemi formlarından ayırt etmek için aşağıdakiler de belirlenir:
MCH (kan sayımına otomatik olarak dahil edilir, normalden yükseğe)
Mikrositik anemi, çok küçük kırmızı kan hücreleri (mikrositler) ile karakterize bir anemidir. Genellikle belirgin veya gizli kan kaybından kaynaklanan bir demir eksikliğini gösterir. Nadir nedenler, doğal ladin gibi ince bağırsak hastalıklarıdır.
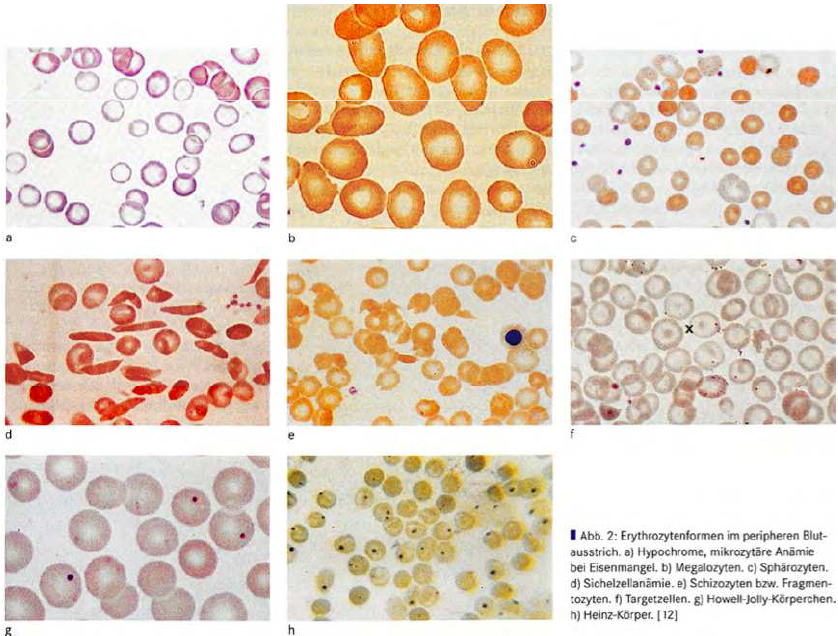
Mikroskopta eritrosit formları: a) demir eksikliğinde görülen hipokrom, mikrositer Anemi; b)Megalositler; c)Spherositler; d) orak hücreli anemi; e)Şizositler; f)Target Hücreleri; g)Howell-Jery- Cisimleri; h)Heinz- Cisimleri
Nedenleri
Kural olarak, mikrositik anemi, demir eksikliğine dayanır. Oksijen bağlayıcı bileşeni (hem) demir gerektiren kırmızı kan pigmenti hemoglobinin işlev bozukluğundan kaynaklanır. Kanama bozukluğu nedeniyle oluşan eritrositler sadece çok küçük olmakla kalmaz, aynı zamanda çok az hemoglobin içerirler ve bu da kan yaymasına indirgenmiş bir renk olarak yansır (hipokromi, hipokromazi). Bu nedenle bir mikrositik anemi genellikle aynı zamanda bir hipokromik anemidir (“mikrositik hipokromik anemi”).
Belirtiler
Semptomlar, kandaki oksijen taşıyıcılarının eksikliğinden kaynaklanan vücuttaki göreceli oksijen eksikliği ile karakterize edilir. Odak noktası – aneminin şiddetine bağlı olarak – kolay yorgunluk, halsizlik, erken bitkinlik, hafif eforla veya zaten istirahat halindeyken nefes darlığı, baş dönmesi ve gözlerin önünde titremedir. Koroner arter hastalığı (koroner yetmezlik, koroner arterlerin daralması) olan kişilerde anemiden dolayı kalp ağrısı (angina pektoris) gelişebilir.
Teşhis
Mikrositik anemide, kan sayımı çok düşük sayıda kırmızı kan hücresi gösterir (eritrosit eksikliği). Kırmızı kan hücreleri de çok küçüktür (düşük MCV, mikrositler) ve çok az hemoglobin içerir (azalmış MCH, azalmış MCHC). Genel olarak, bu düşük bir hematokrite (Hct) yol açar.
Kan sayımında mikrositer anemi saptanırsa, tanı genellikle kanama kaynağı aranarak başlar.
Belirgin bir kanama bulunmazsa (örneğin yaralanma, aşırı adet kanaması (hipermenore)), bir yandan çok hassas olan ve zaten diş etlerinin kanamasını düşündüren dışkıda gizli kan testi (FOBT, dışkıda gizli kan testi) takip eder, Öte yandan, kanamanın tüm olası nedenlerini her zaman kanıtlamaz, böylece kişi yanlış bir güvenlik duygusuna kapılabilir. Gastrointestinal sistem endoskopisi yaşlılarda ve muhtemelen genç insanlarda kesinlikle düşünülmelidir.
Kalın bağırsakta hemorajik inflamasyon (kolit), polipler ve kolon kanseri dışlanmalıdır.
Gastrointestinal sistemin üst kısmında, neden mide mukozasının kanlı bir iltihabı (hemorajik gastrit) veya mide veya duodenumda bir ülser (mide ülseri, duodenum ülseri) olabilir. Koroner arter daralması vakalarında kanı inceltmek için anti-romatizmal ilaçlar veya ASA kullanırken bu nadir değildir.
Kanama kaynağı saptanamazsa, ince bağırsak hastalığından kaynaklanan demir emilim bozukluğu düşünülmelidir. Oldukça nadir görülen bu hastalıklardan çölyak (yerli ladin) göz önünde bulundurulur. Teşhisi için duodenumdan mukoza zarı örneği (duodenumdan biyopsi) ve anti-endomisyal antikorların veya transglutaminazlara karşı antikorların tespiti ile gastroskopi kullanılır.
Terapi
Mikrositer anemi tedavisi, nedene bağlıdır. Demir eksikliği durumunda kanamanın kaynağı belirlenmeli ve tedavi edilmelidir. Bir demir emilim bozukluğu durumunda, enjeksiyon veya infüzyon yoluyla parenteral demir temini gerekli olacaktır. Vücudun zamanla aşırı demir yüklenmesi olmamasına özen gösterilmelidir.
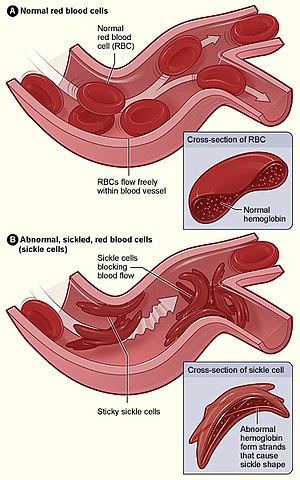
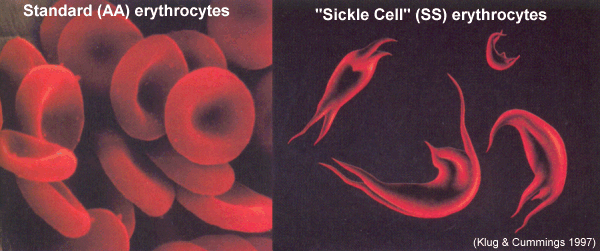
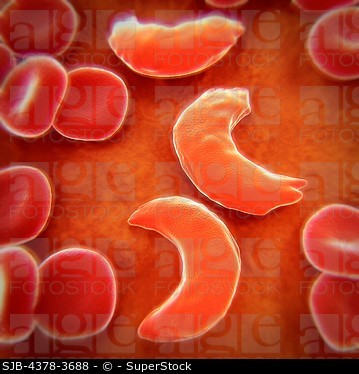
Orak hücreli anemi olarak da bilinir. (Bkz; drepan-o-sit–oz)
Orak hücreli anemi, hemolitik anemilere veya hemoglobinopatilere ait kalıtsal bir hastalıktır. Orak hücre hemoglobini (hemoglobin S, HbS) olarak adlandırılan düzensiz hemoglobin oluşumuna yol açan genetik bir kusur (nokta mutasyonu) tarafından tetiklenir.
Genetik
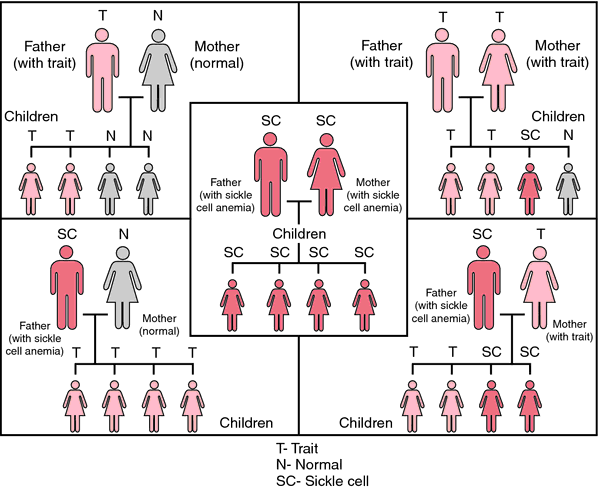
Orak hücre anemisi, otozomal resesif geçişi izler. Pozisyon 6’da hidrofobik valin yerine hidrofilik glutamat ile ß-globin zincirinde bir nokta mutasyonu vardır. HbS’nin oksijensizleştirilmiş durumunda, konsantrasyon yeterince yüksekse agregalar oluşur ve hemoliz meydana gelir.
Yalnızca anne ve baba genlerinin değiştiği özelliğin homozigot taşıyıcıları hastalanır. Heterozigot özellik taşıyıcıları büyük ölçüde semptomsuzdur, çünkü her iki alel de eş-baskın, yani. özelliklerin taşıyıcıları hem normal hem de hatalı hemoglobin üretir. Bununla birlikte, anestezi sırasında olduğu gibi şiddetli oksijen eksikliğinde bile, eritrositlerin orak şekli gelişerek organlara kan akışı bozulabilir. Bileşik heterozigotlar, yani bir HbS aleli ve başka bir patolojik aleli olan hastalar (ör. HbC, HbE), orak hücre anemisine (orak hücre sendromu) benzer bir klinik tablo gösterir.,
Epidemiyoloji
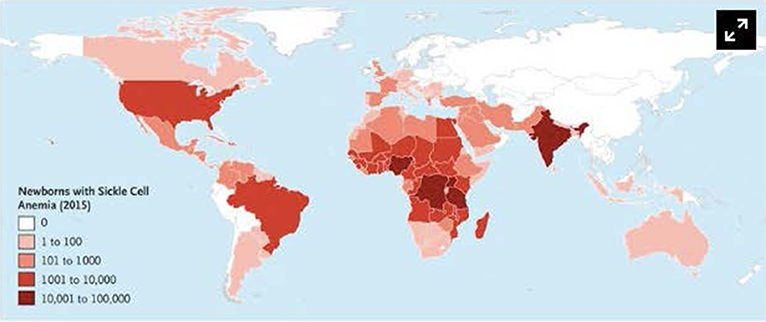
Orak hücre anemisi en çok Afrika ve Asya’nın sıtma bölgelerinde görülür. Ekvator Afrika’da nüfusun% 25-40’ı heterozigot taşıyıcılardır. Ekvatordan uzaklaştıkça kusurun sıklığı önemli ölçüde azalır. Amerika’nın siyah nüfusunda, görülme sıklığı sadece % 5 ile % 10 arasındadır.
Bu fenomen, heterozigot özellik taşıyıcılarının sıtmaya karşı göreceli bir dirence sahip olmasıyla açıklanabilir – bu, sıtma bölgelerinde açık bir seçim avantajını temsil eden bir gerçek. Orta enlemlerde, sıtma eksikliği nedeniyle seçim avantajı etkili değildir.
Almanya’da her yıl yaklaşık 300 çocuk ve yetişkin orak hücre hastalığından etkilenmektedir. Çoğunlukla endemik bölgelerden gelen göçmenlerdir.
Klinik
Orak hücreli anemi genellikle kendini ancak yaşamın 6. ayından itibaren gösterir. Bundan önce, eritrositlerde fetal hemoglobin (HbF) bulunur. Orak hücre anemisinin akut hastalık süreci, akut ağrı ve anksiyete ile ilişkilendirilen orak hücre krizi olarak adlandırılan bir dönemde gerçekleşir. Orak hücre krizlerinin sıklığı ve şiddeti büyük ölçüde değişir. Ağrı herhangi bir yerde olabilir ve birkaç saat ila iki hafta sürebilir.
Tetikleyici
Orak hücre krizlerinin olası tetikleyicileri şunlardır:
Enfeksiyonlar, ateş
Hipoksi (ör. Yüksek rakımda uçarken)
Dehidrasyon
Asidoz
İlaçlarla hemoliz, kontrast madde
Aşırı fiziksel efor
Organ hasarı
Orak hücre anemisindeki vazo-tıkanmalar, tekrarlayan mikro enfarktüslere yol açar ve bu da organ hasarına yol açar. Yaşamın ilk birkaç yılında enfeksiyona duyarlılığı artmış fonksiyonel aspleni ortaya çıkar.
Dalağın akut venöz tıkanması (dalak sekestrasyon krizi) yaşamı tehdit eden bir komplikasyondur.
Orak hücreli anemi’de dalağın otosplenektomiye uğraması sonucu, diğer retikülo endotelyal sistem hücreleri hipertrofiye uğrar, yani adenotonsiller hipertrofiye uğrar.
Retina damarlarının tıkanması kanamalara ve proliferatif retinopatiye neden olabilir.
Renal papiller nekroz, böbrek yetmezliğine kadar varan izostenüriye neden olur. Kemik ve eklem iskemileri aseptik nekroz, kronik artropati ve osteomiyelite yatkınlıkla sonuçlanır.
El-ayak sendromuna parmaklardaki ağrılı kalp krizi neden olur.
Peniste venöz çıkış yolunun enfarktüsü kalıcı iktidarsızlıkla birlikte priapizme yol açabilir.
Alt bacakların kronik ülserasyonları, genellikle ek süperinfeksiyonla birlikte cilt iskemisine dayanır.
Ağrı, taşipne, ateş ve öksürük ile seyreden akut göğüs sendromu, akciğerlerdeki orak hücre krizinin bir ifadesidir.
Azalan arteriyel oksijen satürasyonu, pulmoner hipertansiyon ve kor pulmonale’ye kadar ilerlemeyi destekler. Çocuklarda zaten felç oluyor. MSS’deki kronik subakut hasar, kendini genellikle davranışta ince değişiklikler olarak gösterir.
Tipik anemi semptomlarına ek olarak, hemolitik anemi ayrıca birden fazla komplikasyona (örn. Sarılık, safra taşı) yol açar.
Teşhis
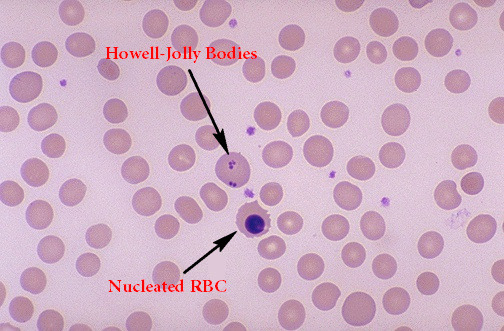
Hemolitik aneminin bir sonucu olarak, hemoglobin konsantrasyonu ve hematokrit önemli ölçüde azalır. Buna genellikle retikülositoz ve granülositoz eşlik eder. Orak hücreler, kan yaymasında ve bazen de dalak fonksiyonunda bozulmanın bir işareti olarak Howell-Jolly cisimlerinde görülebilir.
Orak hücre anemisinin kesin tespiti Hb elektroforezi, kütle spektrometresi ve orak hücre testi ile yapılır. Moleküler genetik test tanıyı doğrulayabilir. Preimplantasyon teşhisi de mümkündür.
Tedavi
Genetik kusuru ortadan kaldıran bir nedensel terapi yoktur.
Hematopoietik kök hücrelere işlevsel bir gen enjekte eden ve hastalığı önemli ölçüde iyileştiren gen terapileri, halihazırda faz II klinik test aşamasındadır.
İşlem, beta talasemide βA-T87Q globin üreten hematopoietik kök hücrelerle tedaviye karşılık gelir. Önümüzdeki birkaç yıl içinde bir pazar lansmanı beklenebilir.
Orak hücre anemisi için halihazırda alınan terapötik önlemler, hastalığın seyrinin ciddiyetine bağlıdır. Diğerlerinin yanı sıra şunları içerir:
Labarotuvarların kullandıkları yöntem, aletlere göre sonuçların değişmesini önlemek adına konulmuş parametrelerdir.
WHo referans Thromobplastini bularak, diğer thromboplastinler sayesinde bulunan Prothrombin ratio’larının karşılaştırılmasını sağlamıştır.Doğrulama Faktörü olarak da ISI yi bulmuştur.
Who tarafından konulan Referans thromboplastinin İsi değeri birdir.bundan dolayı INR=PR’dir.
INR seviyesi nedir?
Kanınızın pıhtılaşmasının ne kadar sürdüğünü kontrol etmek için bir kan testi yaptırdınız. Bu teste PT veya protrombin zamanı testi denir. Testin sonucu INR seviyesi olarak adlandırılır. Varfarin (Coumadin) aldığınızda yüksek bir INR seviyesi oluşabilir. Warfarin kan pıhtılarını önlemeye yardımcı olur.
Kullanım
INR, temel olarak K vitamini antagonistleri (fenprokumon, varfarin) ile antikoagülasyonun seyrini kontrol etmek ve izlemek için kullanılır.
hastalık
INR hedef alan
Derin ven trombozu
2-3
Atriyal fibrilasyon
2-3
Pulmoner emboli
2-3
Mekanik kalp kapakçığı
3-4,5
Hazırlama aşamasında INR, pıhtılaşma önleyici uyum ve stabilite gibi hasta özelliklerine bağlı olarak, ayarlamadan sonra haftada bir ila ayda bir kontrol edilir. Bir INR, kan şekeri testine benzer Coagu Check® gibi sistemler kullanılarak hastanın kendisi tarafından kontrol edilebilir.
Uluslararası Normalleştirilmiş Oran (INR), kanın pıhtılaşma eğilimi için standartlaştırılmış bir ölçüm görevi görür; antikoagülan tedavi gören, özellikle de K vitamini antagonistleri kullanan hastaların yönetimi için çok önemlidir. INR ayarlamaları, kanama veya tromboz riskini en aza indirirken terapötik etkinliğin sürdürülmesinde çok önemlidir. Yüksek INR değerleri, olumsuz sonuçları hafifletmek için dozaj ayarlamaları veya diyet müdahaleleri gerektiren yüksek kanama riskini gösterir. Tersine, düşük INR düzeyleri bireyleri trombotik olaylara yatkın hale getirebilir, bu da INR değerlerini etkileyen faktörlerin titizlikle yönetilmesinin gerekliliğini vurgular. Bu, diyetle alınan K vitamini alımını, ilaç etkileşimlerini ve diğer eksojen faktörleri kapsar. INR seviyelerinin stratejik yönetimi, hasta sonuçlarını optimize etmek için bu değişkenlerin kapsamlı bir şekilde anlaşılmasını içerir.
INR sistemi, özellikle varfarin gibi K vitamini antagonistleriyle antikoagülasyon tedavisinin izlenmesi ve yönetilmesinin ayrılmaz bir parçasıdır. INR, kanın pıhtılaşma süresinin ölçümünü standartlaştırarak, sağlık hizmeti sağlayıcılarının antikoagülan dozlarını hasta kanını terapötik bir aralıkta tutacak şekilde ayarlamasına olanak tanır ve böylece antikoagülasyonun az veya fazla olmasıyla ilişkili komplikasyonları önler.
Yüksek INR: Riskler ve Yönetim
Yüksek INR, pıhtılaşma süresinin uzadığını gösterir ve hastaları artan kanama riskine sokar. Bu, antikoagülanların dozunu ayarlayarak veya INR’nin önemli ölçüde yüksek olduğu durumlarda, antikoagülasyon etkisini ortadan kaldırmak için K vitamini uygulanarak yönetilebilir. Tedavi stratejisi INR yüksekliğinin düzeyine, kanama varlığına ve hastanın genel tromboz riskine bağlıdır.
Düşük INR: Etkiler ve Düzenlemeler
Tersine, düşük bir INR, daha hızlı bir pıhtılaşma süresine işaret eder ve felç veya derin ven trombozu gibi trombotik olay riskini artırır. Yönetim tipik olarak antikoagülan ilaç dozunun arttırılmasını içerir. Hastalara ayrıca INR seviyelerindeki dalgalanmaları önlemek için diyetle K vitamini alımını izlemeleri ve tutarlı bir şekilde sürdürmeleri önerilir.
INR Düzeylerini Etkileyen Faktörler
Aşağıdakiler de dahil olmak üzere çeşitli faktörler INR seviyelerini etkileyebilir:
Diyetle K Vitamini Alımı: K vitamini açısından yüksek gıdalar (örneğin yeşil yapraklı sebzeler) INR’yi düşürebilirken, K vitamini alımındaki azalma INR’yi artırabilir.
İlaç Etkileşimleri: Bazı ilaçlar warfarinin etkisini artırabilir veya azaltabilir, bu da INR seviyelerinde dalgalanmalara neden olabilir.
Bitki ve Takviye Etkileşimleri: Bazı şifalı bitkiler ve takviyeler, warfarin metabolizmasını veya K vitamini durumunu etkileyerek INR’yi etkileyebilir.
Alkol Tüketimi: Aşırı veya tutarsız alkol alımı INR düzeylerini değiştirebilir.
Sağlık Durumu: Pıhtılaşma faktörü üretimini etkileyen karaciğer fonksiyonu INR seviyelerini etkileyebilir. K vitamini emilimini veya metabolizmasını etkileyen koşullar da INR’yi etkiler.
Tedavinin İzlenmesi ve Ayarlanması
K vitamini antagonist tedavisi gören hastalar için INR düzeylerinin düzenli olarak izlenmesi çok önemlidir. İzleme sıklığı, INR değerlerinin stabilitesine ve hastanın ilaç rejiminde, diyetinde veya sağlık durumundaki değişikliklere bağlı olarak değişebilir. Antikoagülan dozajında veya diyet tavsiyesinde ayarlamalar, trombozu önleme ile aşırı kanamayı önleme arasındaki dengeyi korumak için INR okumalarına göre yapılır.
Sonuç olarak, INR düzeylerinin etkili yönetimi, ilaç dozajı, diyet yönetimi ve diğer maddelerle etkileşimlerin veya sağlık durumundaki değişikliklerin izlenmesinin karmaşık bir etkileşimidir. Bu, güvenli ve etkili antikoagülasyon tedavisi sağlamak için hasta eğitiminin ve sağlık hizmeti sağlayıcılarıyla düzenli iletişimin önemini vurgulamaktadır.
Keşif ve Tarihsel Arkaplan
INR kavramı, kanın pıhtılaşması için geçen süreyi ölçen Protrombin Süresi (PT) testini standartlaştırma ihtiyacından ortaya çıktı. PT testinin duyarlılığı, kullanılan tromboplastin reaktiflerindeki farklılıklar nedeniyle farklı laboratuvarlar arasında önemli ölçüde farklılık gösterdi. Bu değişkenlik, klinisyenlerin farklı laboratuvarlardan alınan PT sonuçlarını güvenilir bir şekilde karşılaştıramaması nedeniyle, varfarin tedavisi gören hastalarda tutarlı antikoagülasyon seviyelerinin korunmasında zorluklara yol açtı.
1980’lerde Dünya Sağlık Örgütü (WHO) standardizasyonun gerekliliğini fark etti ve bu konuyu ele almak için INR sistemini uygulamaya koydu. INR sistemi, kullanılan tromboplastin reaktiflerine bakılmaksızın PT sonuçlarını standartlaştırmak için Uluslararası Hassasiyet İndeksini (ISI) kullanan bir kalibrasyon modeline dayanıyordu.
Modern Katkılar ve Etki
Laboratuvarlar Arasında Standardizasyon: INR’nin en önemli katkısı, dünya çapındaki farklı laboratuvarlarda PT testlerinin uyumlaştırılmasındaki rolüdür. Bu standardizasyon, antikoagülasyon tedavisinin tutarlı ve doğru şekilde izlenmesini kolaylaştırarak hasta güvenliğini artırdı.
Antikoagülasyon Tedavisinin Yönetimi: INR, warfarin kullanan hastaların tedavisinde devrim yaratarak dozajların daha hassas bir şekilde ayarlanmasına olanak tanıdı. Bu, yetersiz veya aşırı antikoagülasyonla ilişkili komplikasyon riskini önemli ölçüde azaltmıştır.
Kılavuz Geliştirme: INR’nin kullanıma sunulması, antikoagülasyon tedavisi gerektiren çeşitli durumlar için hedef INR aralıkları için net öneriler sağlayan klinik kılavuzların geliştirilmesine yol açmıştır. Bu kılavuzlar hasta sonuçlarının iyileştirilmesinde etkili olmuştur.
Araştırma ve Geliştirme: Standartlaştırılmış INR sistemi aynı zamanda yeni antikoagülan ilaç ve tedavilere yönelik araştırmaların yolunu da açmıştır. Yeni antikoagülanların etkinliğini ve güvenliğini değerlendiren klinik çalışmalarda temel bir araç haline geldi.
Küresel Sağlık Etkisi: INR’nin standardizasyonu, küresel sağlık üzerinde derin bir etki yaratarak, çeşitli sağlık hizmeti ortamlarındaki hastaların tutarlı ve etkili antikoagülasyon yönetimi almasını sağladı. Bu, dünya çapında milyonlarca hastanın bakım kalitesini artırdı.
İleri Okuma
Holbrook, A., Schulman, S., Witt, D. M., Vandvik, P. O., Fish, J., Kovacs, M. J., Svensson, P. J., Veenstra, D. L., Crowther, M., & Guyatt, G. H. (2012). Evidence-based management of anticoagulant therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest, 141(2 Suppl), e152S-e184S.
Ansell, J., Hirsh, J., Hylek, E., Jacobson, A., Crowther, M., & Palareti, G. (2008). Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest, 133(6_suppl), 160S-198S.
Favaloro, E. J. (2008). Clinical application of the INR: Review and recommendations for laboratory monitoring of oral anticoagulant therapy.Haemophilia, 14(5), 929-942.
Keeling, D., Baglin, T., Tait, C., Watson, H., Perry, D., Baglin, C., Kitchen, S., & Makris, M. (2011). Guidelines on oral anticoagulation with warfarin – fourth edition. British Journal of Haematology, 154(3), 311-324.
Tripodi, A. (2011). The International Normalized Ratio (INR): A guide to understanding and correcting its problems. Clinical Chemistry and Laboratory Medicine, 49(4), 651-656.
Kitchens, C. S., Erkan, D., Brandão, L. R., Hylek, E., Jacobsen, A. F., Pollack, C. V., Spyropoulos, A. C., Stafford-Smith, M., & Douketis, J. D. (2012). Consultative hemostasis and thrombosis. Saunders Elsevier.
Wittkowsky, A. K. (2004). Warfarin and other coumarin derivatives: Pharmacokinetics, pharmacodynamics, and drug interactions. Seminars in Vascular Medicine, 4(3), 271-280.
Gage, B. F., & Fihn, S. D. (2003). Management of anticoagulation therapy for patients receiving warfarin. Journal of Thrombosis and Thrombolysis, 15(1), 31-41.
Hirsh, J., Dalen, J., Anderson, D. R., Poller, L., Bussey, H., Ansell, J., & Deykin, D. (2001). Oral anticoagulants: Mechanism of action, clinical effectiveness, and optimal therapeutic range.Chest, 119(1_suppl), 8S-21S.
Poller, L., & Keown, M. (1988). The Standardization of the Prothrombin Time for the International Normalized Ratio System: A Re-Examination of the Concept and Its Application. Journal of Thrombosis and Haemostasis, 60(1), 1-8.
Tripodi, A. (2011). The International Normalized Ratio (INR) calibration model: The key to improving sensitivity and reliability in oral anticoagulant control. Thrombosis Research, 127(Suppl 3), S8-S11.
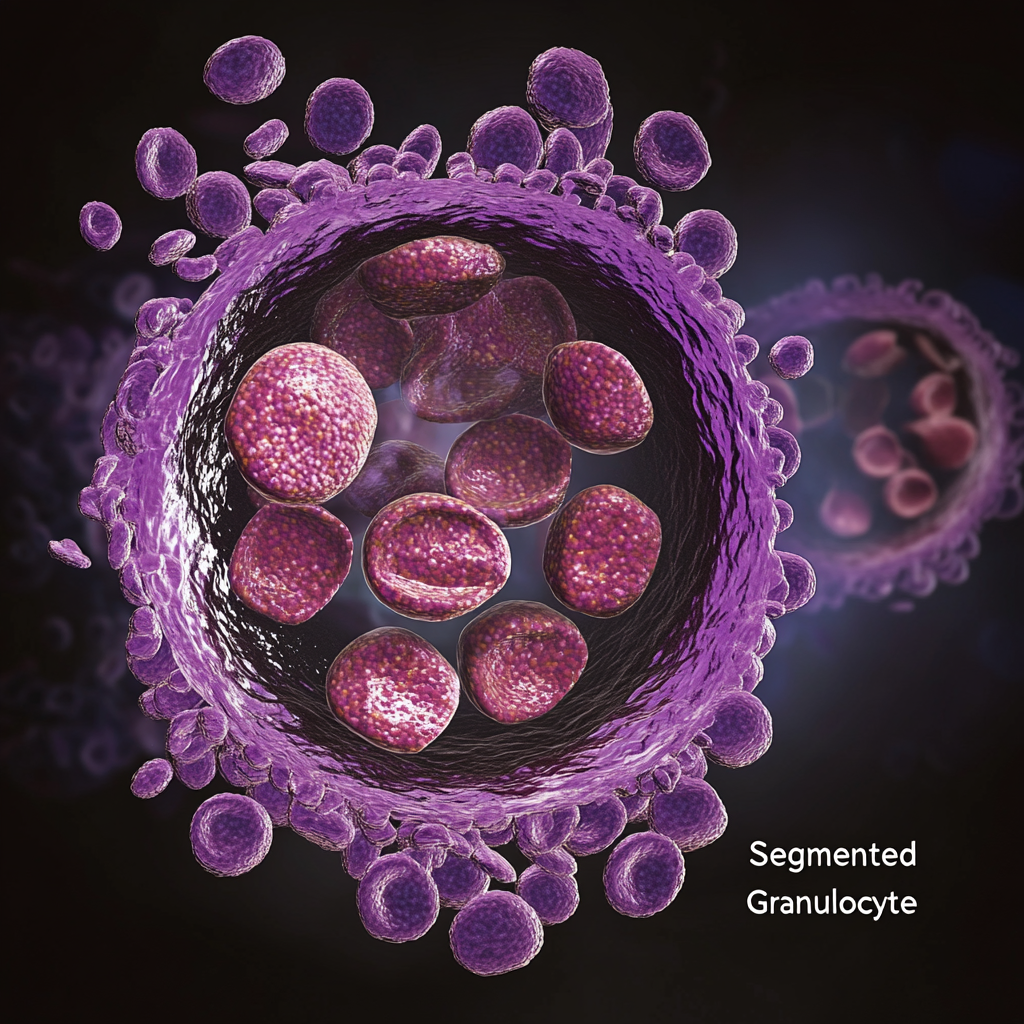
“Segmentli granülosit” terimi bu hücrelerin iki temel özelliğinden türetilmiştir:
“Segmentli” olgun nötrofillerdeki çekirdeğin ayırt edici şeklini ifade eder. Çekirdek 2-5 lob veya segmente bölünerek polimorfonükleer bir görünüm kazanır. Bu segmentasyon nötrofil olgunlaşmasının son aşamalarında, çekirdeğin bant şeklinden tamamen segmentli bir forma geçişiyle gerçekleşir.
“Granülosit”, hücrenin sitoplazmasında spesifik granüllerin varlığını tanımlar. Bu granüller çeşitli antimikrobiyal ajanlar, enzimler ve hücrenin bağışıklık fonksiyonu için çok önemli olan diğer maddeleri içerir.
“Nötrofil” teriminin kendisi hücrenin boyanma özelliklerinden gelmektedir. Hematoksilen ve eozin (H&E) boyalarına maruz kaldıklarında nötrofiller, bazofillerin koyu mavisi veya eozinofillerin parlak kırmızısının aksine nötr pembe bir renk alır.
Bu nedenle, “segmentli granülosit” tam terimi, hücrenin ayırt edici nükleer morfolojisini ve sitoplazmik granüllerin varlığını doğru bir şekilde tanımlar; her ikisi de olgun nötrofillerin tanımlayıcı özellikleridir.
Bölümlü Granülositler
Granülopoezde Tanım ve Rol Bölümlü granülositler, polimorfonükleer lökositler (PMN’ler) olarak da bilinir, granülosit gelişiminin (granülopoez) son, olgun aşamasını temsil eder. İnce kromatin filamentleriyle birbirine bağlanan 2-5 ayrı loba bölünmüş bir çekirdek ile karakterize edilirler. Bu hücreler periferik kanda en bol bulunan granülositlerdir ve toplam lökositlerin %45-70’ini oluştururlar, nötrofiller baskın alt tiptir (dolaşımdaki bölümlü granülositlerin ~%95’i).
This content is available to members only. Please login or register to view this area.
Gelişimsel Bağlam (Granülopoez) Granülopoez kemik iliğinde meydana gelir ve şu aşamalardan geçer:
Bölümlü granülositler “bant hücresi” aşamasını (çubuk çekirdekli) takip eder ve dolaşıma salınır.
G-CSF (Granülosit Koloni Uyarıcı Faktör) gibi sitokinler tarafından düzenlenen bu süreç ~10–14 gün sürer.
Yaşam süresi: Dokulara göç etmeden önce 6–10 saat dolaşımda kalır ve 1–4 gün yaşar.
Morfoloji
Boyut: 9–16 μm çapında.
Çekirdek:
2–5 lob (ortalama: 3), nükleer filamentlerle birbirine bağlanmıştır.
Kromatin, olgunlaşma nedeniyle yoğun bir şekilde kümelenmiştir (piknotik).
Sitoplazma:
Nötrofiller: Enzimler (örn. alkalin fosfataz, lizozim) ve antimikrobiyal proteinler içeren ince, soluk pembe granüller (spesifik/ikincil granüller). Gelişimin erken dönemlerinde oluşan azurofilik (birincil) granüller devam eder.
Eozinofiller: Büyük, parlak turuncu-kırmızı granüller (ana bazik protein açısından zengin).
Bazofiller: Kaba, koyu mavi granüller (heparin, histamin içerir).
Histolojik Tanımlama
Boyama: Periferik kan yaymalarında Wright-Giemsa boyası.
Temel Özellikler:
Nötrofiller: Soluk sitoplazmaya sahip çok loblu çekirdek (Şekil 1A).
Eozinofiller: Belirgin turuncu granüllere sahip iki loblu çekirdek.
Bazofiller: Yoğun mavi granüller nedeniyle belirsiz çekirdek.
Hipersegmentasyon: ≥5 nükleer lob, B12 vitamini/folat eksikliği veya yaşlanmayla ilişkilidir.
Klinik Önem
Enfeksiyon/İltihaplanma:
Bakteriyel enfeksiyonlarda artan nötrofiller (nötrofili).
“Sola kayma”: Şiddetli enfeksiyonlarda olgunlaşmamış formların (bantlar) varlığı.
Tam Kan Sayımı (CBC): Granülosit alt kümelerini niceliksel olarak belirler.
Ayırıcı Yayma: Nükleer segmentasyonu, granülasyon desenlerini ve anormallikleri (örneğin sepsiste toksik granüller) belirler.
Patolojik Hususlar
Reaktif Değişiklikler: Toksik granülasyon, Döhle cisimcikleri (mavi sitoplazmik inklüzyonlar) veya şiddetli enfeksiyonlarda vakuolleşme.
Displastik Değişiklikler: Miyelodisplastik sendromlarda hipogranülarite veya psödo-Pelger-Huët anomalisi (iki loblu çekirdekler).
Şekil Referansları
Bölümlü Granülositler için Şekil Referansları Bölümlü granülositlerin görsel olarak tanımlanması için, şu standart histoloji/hematoloji kaynaklarına bakın:
Hematoloji Atlası (WHO Sınıflandırması)
Şekil: Bölümlü çekirdeğe sahip nötrofil (genellikle granülosit olgunlaşması üzerine bölümlerde etiketlenmiştir).
Kaynak: Kumar & Clark’ın Klinik Tıp veya Robbins Temel Patoloji.
Wright-Giemsa Boyalı Kan Yayması Görüntüleri
Çevrimiçi Veritabanları:
CellWiki – “Bölümlü nötrofil” veya “polimorfonükleer lökosit” ifadesini arayın.
Wikimedia Commons – Kan hücrelerinin yüksek çözünürlüklü görüntüleri.
Hipersegmente Nötrofil: ≥5 nükleer lob (megaloblastik anemide görülür).
Normal segmentasyonla karşılaştırın (2–5 lob).
Boyama Sorun Giderme
Aşırı renk açma: Çekirdek soluk görünür; durulama süresini azaltın.
Az boyama: Granüller görünmez; boyama süresini artırın.
Çökeltiler: Kullanmadan önce boyayı filtreleyin.
Mikroskobik Değerlendirme İçin Önemli Notlar
Bant ve Segmentli Nötrofil:
Bant hücre: Çekirdek U şeklindedir ve daralma yoktur.
Bant hücre: Filamentlerle birbirine bağlı loblara belirgin bir şekilde ayrılmıştır.
Patolojik Bulgular:
Toksik Granülasyon: Koyu renkli iri granüller (sepsis, inflamasyon).
Döhle Cisimcikleri: Mavi sitoplazmik inklüzyonlar (enfeksiyon, yanıklar).
Keşif
Bölümlere ayrılmış granülositlerin keşfi, 19. yüzyılın sonlarında mikroskopi ve boyama tekniklerindeki ilerlemelerle başlamıştır. Alman bir doktor ve bilim insanı olan Paul Ehrlich, kömür katranı boyaları kullanarak kan hücrelerini ayırt etmek için yöntemler geliştirerek çok önemli bir rol oynamıştır. 1879-1880 yılları arasında yayınlanan çalışması, nötrofiller de dahil olmak üzere çeşitli lökositlerin tanımlanmasına olanak sağlamıştır. Ehrlich bu hücreleri “polimorf çekirdekli hücreler” olarak tanımlamış, parçalı çekirdeklerine ve nötr olarak boyanan granüllerin varlığına dikkat çekmiştir. Bu, nötrofillerin ayrı bir hücre tipi olarak ilk kez net bir şekilde tanınmasına işaret ederek hematoloji ve immünolojinin temelini attı.
Ehrlich’in teknikleri sadece nötrofilleri tanımlamak için değil, aynı zamanda lösemiler ve anemiler gibi kan hastalıklarının sınıflandırılmasını sağlamak için de önemliydi. Yaklaşımı, hücre tiplerini ayırt etmek için asidik, bazik ve nötr boyalar kullanmayı içeriyordu ki bu, doktora tezinde mast hücrelerini keşfetmesinin doğrudan bir devamıydı. Bu çalışma, Paul Ehrlich Wikipedia adresinde bulunanlar gibi tarihsel anlatımlarda ayrıntılı olarak açıklanmıştır.
İşlevsel İçgörüler: Fagositozun Rolü
Ehrlich’in morfolojik tanımlamasının ardından, nötrofillerin işlevsel rolü Élie Metchnikoff’un çalışmalarıyla ortaya çıkmaya başlamıştır. 1884 yılında Rus bir biyolog olan Metchnikoff, hücrelerin bakteri gibi yabancı partikülleri yuttuğu ve sindirdiği bir süreç olan fagositozu tanımlamıştır. Gözlemleri, o zamanlar özel olarak adlandırmamış olsa da, şu anda nötrofil olarak tanıdığımız hücreleri içeriyordu. Metchnikoff’un çalışması, fagositozu temel bir bağışıklık mekanizması olarak ortaya koyması ve nötrofillerin de bu sürece dahil olan birincil hücreler arasında yer alması nedeniyle çığır açıcı olmuştur. Bu durum, Metchnikoff’un 1880’lerde “fagosit” terimini ortaya attığını belirten Phagocytosis Britannica gibi kaynaklarda belirtilmektedir.
Metchnikoff’un keşfi, daha sonra kendisine Ehrlich ile birlikte 1908 Nobel Fizyoloji veya Tıp Ödülü’nü kazandıran doğuştan gelen bağışıklık üzerine yaptığı daha geniş araştırmanın bir parçasıydı. Frontiers Immunology] (https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2012.00174/full) gibi çalışmalarda belirtildiği üzere, gözlemleri nötrofillerin mikroorganizmalara karşı “başlıca savaşçılar” olduğunu vurgulamıştır.
Resmi Tanınma ve Nobel Ödülü
Nötrofillerin bağışıklık sistemindeki önemi, 1908 yılında Paul Ehrlich ve Élie Metchnikoff’un bağışıklık konusundaki çalışmaları nedeniyle Nobel Fizyoloji veya Tıp Ödülü’ne layık görülmesiyle resmen tanınmıştır. Ehrlich’in katkıları arasında beyaz kan hücrelerinin tanımlanması ve sınıflandırılması yer alırken, Metchnikoff’un fagositoz üzerine yaptığı çalışmalar nötrofillerin işlevsel rolünü anlamamızı sağlamıştır. Bu dönüm noktası, Nobel Ödülü Ehrlich ve Nobel Ödülü Metchnikoff‘da belgelendiği üzere, nötrofillerin immünolojik araştırmalardaki yerini sağlamlaştırmıştır.
Sonraki Gelişmeler ve Araştırmalar
İlk keşif ve işlevsel anlayış 19. yüzyılın sonları ve 20. yüzyılın başlarında yerleşmiş olsa da, nötrofiller üzerine yapılan araştırmalar gelişmeye devam etmiştir. 1930’larda, nötrofillerdeki solunum patlaması keşfedilmiştir; bu, mikrobisidal aktiviteleri için çok önemli olan fagositoz sırasında artan oksijen tüketimini içeren bir süreçtir. Bu durum ASH Blood Phagocytes gibi tarihsel incelemelerde belirtilmiştir.
yüzyılın ortalarında, mikroskopi ve biyokimyadaki gelişmeler, spesifik granüllerin ve enzimler ve antimikrobiyal proteinler gibi içeriklerinin tanımlanması da dahil olmak üzere nötrofil yapısının daha derinlemesine anlaşılmasını sağlamıştır. 20. yüzyılın sonlarında, PMC Neutrophil Advances gibi modern incelemelerde tartışıldığı gibi, nötrofil aktivasyonu ve fagositozda yer alan spesifik reseptörleri ve sinyal yollarını ortaya çıkaran moleküler biyoloji tekniklerinin uygulanmasına tanık oldu.
yüzyılda yapılan araştırmalar, nötrofil fonksiyonunun karmaşıklığını ortaya çıkarmış, enflamasyon, immün modülasyon ve kanser ve otoimmün bozukluklar gibi hastalık patogenezindeki rollerini ortaya koymuştur. Bu durum Nature Neutrophil Diversity gibi son çalışmalarda ayrıntılı olarak açıklanmaktadır.
İleri Okuma
Ehrlich, P. (1879). Beiträge zur Kenntniss der Anilinfärbungen und ihrer Verwendung in der mikroskopischen Technik. Archiv für mikroskopische Anatomie, 16, 263–277.
Ehrlich, P. (1880). Methodologische Beiträge zur Physiologie und Pathologie der verschiedenen Formen der Leukocyten. Zeitschrift für klinische Medizin, 1, 553–606.
Schridde, H. (1908). Über die Entwicklung und Bedeutung der neutrophilen Leukocyten im menschlichen Organismus. Virchows Archiv für pathologische Anatomie und Physiologie und für klinische Medizin, 191, 363–408.
Maximow, A. A. (1909). Über die Morphologische Grundlage der Blutbildenden Gewebe und ihre Bedeutung für die Frage der Blutbildung. Folia Haematologica, 8, 125–199.
Arneth, J. (1904). Über die quantitative Untersuchung der verschiedenen kernigen Formen der neutrophilen Leukocyten im normalen Blut und bei Leukämie. Zeitschrift für klinische Medizin, 54, 511–534.
Schilling, V. (1924). Das Verhalten der neutrophilen Kernformen bei Infektionskrankheiten. Zeitschrift für die gesamte experimentelle Medizin, 41, 436–458.
Fliedner, T. M., & Steinbach, K. (1958). Zellkinetische Untersuchungen über die Entwicklung segmentkerniger Granulozyten. Blut, 4(4), 202–218.
Granülosit olgunlaşmasının çubuk şeklinde, parçalanmamış bir çekirdekle karakterize edilen bir aşamasıdır. (bkz: granulosit )
Granülopoez
Çubuk çekirdekli granülositler granülopoezin sondan bir önceki aşamasıdır. Granülopoez sıralamasında metamiyelositlerden sonra ve segment çekirdekli granülositlerden önce yer alırlar ve hem kemik iliğinde hem de periferik kanda bulunurlar. Burada, çubuk çekirdekli granülositler normalde lökositlerin %3’üne kadar bir paya sahip olabilir.
Morfoloji
Çubuk çekirdekli granülositlerin çekirdeği parçalanmamış ve at nalı şeklinde veya S kıvrımlıdır, bu da bu hücre formuna adını verir. Sitoplazma spesifik granülasyonlar içerir.
Klinik
Çubuk çekirdekli granülositlerin artış göstermesi, diferansiyel kan tablosunda sola kayma olarak adlandırılan durumun bir özelliğidir ve bir enfeksiyonun varlığına işaret eder.
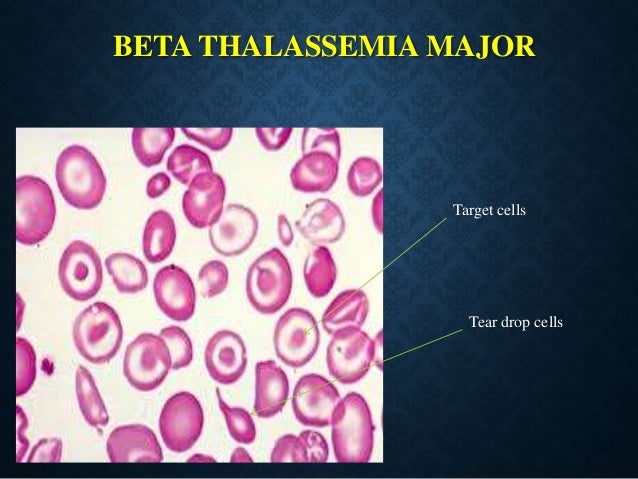
Talasemi, Yunanca “thalassa ‘ (’deniz” anlamına gelir) ve “anemi ” (kan eksikliği veya yetersiz kırmızı kan hücresi anlamına gelir) kelimelerinden kaynaklanan genetik bir hemoglobin üretim bozukluğudur. Bu isim, hastalığın Akdeniz toplumlarında, özellikle de denize yakın bölgelerde yüksek prevalansını yansıtmaktadır. Talasemi, hemoglobin molekülünü oluşturan protein zincirlerinden birinde (alfa veya beta) eksiklikle karakterize olup, asemptomatikten şiddetli, hayatı tehdit eden anemiye kadar çeşitli klinik belirtilere yol açar.
Talaseminin Etimolojisi
Talasemi** terimi 20. yüzyılın başlarında Yunanca’dan türetilmiştir:
Thalassa**: “Deniz” anlamına gelir ve bu durumun ilk kez tanımlandığı Akdeniz bölgesine atıfta bulunur.
Anemi**: Durumun ayırt edici özelliği olan yeterli kırmızı kan hücresi veya hemoglobin eksikliğini ifade eder.
Talasemi Epidemiyolojisi
Talasemi özellikle tropikal ve subtropikal bölgelerde, özellikle de tarihsel olarak sıtmaya maruz kalmış popülasyonlarda yaygındır. Bu durum Akdeniz, Afrika, Orta Doğu, Hint Yarımadası ve Güneydoğu Asya’da oldukça yaygındır. Bu bölgelerdeki yaygınlığının nedeni, talasemi taşıyıcılarının sıtmaya karşı bir miktar korumaya sahip olduğu ve bu popülasyonlarda özelliğin daha yüksek bir sıklığa yol açtığı sıtma hipotezi ile ilgilidir.
Küresel yaygınlık**: Küresel nüfusun yaklaşık *%1,5’i* (90 milyondan** fazla kişi) β-talasemi özelliği taşımaktadır.
Ağır vakaların doğum oranı**: Her yıl dünya çapında *60.000 ila 80.000 ağır talasemi vakasının* doğduğu tahmin edilmektedir.
Talaseminin Klinik Belirtileri
Talasemi, şiddetine bağlı olarak farklı şekillerde ortaya çıkar. Hastalık klinik tabloya göre üç ana forma ayrılır:
Talasemi Minör (Kalıtsal):
Genellikle asemptomatik veya hafif anemi ile seyreder.
Bireyler taşıyıcıdır ancak tipik olarak tedavi gerektirmezler.
Talasemi Major (Cooley Anemisi):
Bebeklik döneminde başlayan şiddetli anemi.
Belirtiler arasında büyüme ve gelişmede gecikme, iskelet anormallikleri (özellikle yüz ve kafatasında), dalak büyümesi ve sarılık yer alır.
Hastalar, transfüzyonlardan kaynaklanan aşırı demir yükünü yönetmek için ömür boyu kan transfüzyonu ve şelasyon tedavisi gerektirir.
Talasemi İntermedia:
Semptomlar talasemi minör ve majör arasında orta şiddette seyreder.
Bu bireylerde hafif ila şiddetli anemi, büyümede yavaşlama ve kemik anormallikleri olabilir.
Tedavi semptomların ciddiyetine bağlıdır ve ara sıra kan nakli ve demir şelasyon tedavisi içerebilir.
Talasemi Türleri
Talasemi, etkilenen globin zincirine (alfa veya beta) ve ilgili gen mutasyonlarının sayısına göre sınıflandırılır.
Alfa Talasemi
Alfa talasemi, hemoglobinin alfa globin zincirlerini kodlayan HBA1 ve HBA2 genlerindeki mutasyonlardan kaynaklanır. Kusurlu genlerin sayısına göre sınıflandırılan dört tip alfa talasemi vardır:
Sessiz Taşıyıcı: Bir kusurlu alfa geni; semptom yok.
Alfa Talasemi Minör (Özellik): İki kusurlu alfa geni; hafif anemi.
Hemoglobin H Hastalığı: Üç kusurlu alfa geni; orta ila şiddetli anemi, dalak büyümesi ve kemik deformiteleri.
Alfa Talasemi Major (Hidrops Fetalis): Dört kusurlu alfa geni; bu durum tedavi edilmezse rahimde veya doğumdan kısa bir süre sonra ölümcüldür.
Beta Talasemi
Beta talasemi, hemoglobinin beta globin zincirini kodlayan HBB genindeki mutasyonlardan kaynaklanır. İki ana formu vardır:
Beta Talasemi Major (Cooley Anemisi): Beta globin üretiminin tamamen olmaması şiddetli anemiye yol açar. Hastalar ömür boyu kan nakline ve şelasyon tedavisine ihtiyaç duyar.
Beta Talasemi Minör (Özellik): Beta globin üretiminin azalması, genellikle tedavi gerektirmeyen hafif anemiye yol açar.
Alfa ve beta talasemiye ek olarak, hemoglobinin delta ve gama zincirlerini etkileyen delta ve gamma talasemi gibi nadir formlar da vardır. Bu formlar da spesifik genlerdeki mutasyonları içerir ve genetik testler ve laboratuvar yöntemleri kullanılarak teşhis edilir.
Global Tanı Kriterleri
Talasemi tanısı tipik olarak aşağıdaki testleri içerir:
Tam Kan Sayımı (CBC)**: Anemi ve kırmızı kan hücresi anormalliklerini kontrol etmek için.
Hemoglobin Elektroforezi**: Beta talasemide yüksek fetal hemoglobin (HbF) ve hemoglobin A2 (HbA2) seviyeleri gibi mevcut hemoglobin türlerini ve miktarlarını belirlemek için.
DNA Analizi**: Kesin tanı için, alfa veya beta globin genlerindeki mutasyonları veya delesyonları tanımlamak üzere genetik testler kullanılır.
Tedavi Kılavuzları
Talaseminin tedavisi ciddiyetine bağlıdır:
Talasemi Minör:
Genellikle tedavi gerektirmez. Hastalar tipik olarak asemptomatiktir ve sadece hafif anemileri olabilir.
Talasemi Major:
Düzenli kan transfüzyonları**: Hemoglobin seviyelerini korumak ve normal büyüme ve gelişmeyi sağlamak için gereklidir.
Demir şelasyon tedavisi**: Düzenli kan nakillerinden kaynaklanan aşırı demir yüklenmesini önlemek için verilir. Fazla demirin vücuttan atılması için *deferoksamin* veya deferasiroks gibi ilaçlar kullanılır.
Folik asit takviyeleri**: Kırmızı kan hücresi üretimini desteklemek için önerilebilir.
Kemik iliği veya kök hücre nakli**: Bu, uygun bir donör mevcutsa şiddetli talasemi için tek potansiyel tedavidir.
Talasemi İntermedia:
Tedavi semptomların şiddetine bağlı olarak değişir ancak ara sıra kan transfüzyonu ve demir şelasyon tedavisini içerebilir.
Son Gelişmeler ve Gelecekteki Yönelimler
Gen Terapisi: Talasemi için gen terapisi araştırmaları, altta yatan genetik kusuru düzeltmek ve potansiyel olarak hastalığı iyileştirmek amacıyla devam etmektedir.
Geliştirilmiş Demir Şelasyon Tedavileri**: Şelasyon ilaçlarındaki gelişmeler, aşırı demir yükünün yükünü azaltarak hastaların yaşam kalitesini artırmıştır.
Doğum Öncesi Test ve Genetik Danışmanlık**: Talasemi prevalansının yüksek olduğu bölgelerde, doğum öncesi genetik test ve danışmanlık, hastalığın erken teşhisi ve yönetimi için önemlidir.
Keşif
1904 – Talaseminin İlk Tanımı
Yunan doktor Thomas Bizzozero şiddetli anemi ve kemik deformiteleri olan çocuk vakalarını belgeleyerek talaseminin bilinen ilk tanımını yaptı.
1925 – Talaseminin İsimlendirilmesi
Dr. George Whipple ve Dr. Thomas Cooley Akdeniz kökenli çocuklarda ciddi bir anemi türü tespit etmiştir. Daha sonra beta-talasemi major olarak bilinen Cooley anemisine Dr. Cooley’in adı verilmiştir.
1932 – “Talasemi” Terimi Ortaya Çıktı
Talasemi** terimi Dr. George Whipple tarafından ortaya atılmış olup, Yunanca “thalassa” (deniz) ve “anemi” kelimelerinden türetilmiştir ve hastalığın Akdeniz toplumlarındaki yüksek prevalansını yansıtmaktadır.
1949 – Talasemi için Kan Transfüzyonu
Talasemi majör tedavisi olarak düzenli kan transfüzyonlarının ilk kullanımı başlatıldı ve şiddetli anemi yönetiminde bir köşe taşı haline geldi.
1960’lar – Demir Şelasyon Tedavisinin Geliştirilmesi
Sık kan transfüzyonlarından kaynaklanan aşırı demir yükünü tedavi etmek için Demir şelasyon tedavisi kullanılmaya başlandı. Bu, talasemi hastalarının uzun vadeli yönetiminde önemli bir gelişmeye işaret ediyordu.
1970’ler – Talasemide Fetal Hemoglobinin Keşfi
Araştırmacılar fetal hemoglobin (HbF) üretiminin beta-talasemide semptomların şiddetini azaltabileceğini keşfederek HbF seviyelerini artırmaya yönelik terapötik stratejilerde ilerlemelere yol açtı.
1980’ler – Prenatal Tanı ve Genetik Danışmanlık
Genetik testlerin kullanıldığı Prenatal tanı geliştirilerek fetüslerde talaseminin erken teşhisi sağlandı ve aile planlaması ile bilinçli karar verme süreçlerine olanak tanındı.
2000’ler – Gen Terapisi Araştırmaları
Altta yatan genetik kusuru düzelterek talasemiyi iyileştirmeyi amaçlayan gen terapisi araştırmalarındaki ilerlemeler, gelecekteki iyileştirici tedaviler için umut vaat ediyor.
2010’lar – Geliştirilmiş Şelasyon İlaçları
Önceki tedavilere kıyasla daha kolay oral uygulama ve daha iyi uyum sunan deferasiroks gibi yeni demir şelatlama ajanları geliştirildi.
2019 – Beta-Talasemi için Gen Tedavisinin Onaylanması
Avrupa İlaç Ajansı, beta-talasemi hastaları için bir gen terapisi olan Zynteglo‘yu onaylayarak hastalığın iyileştirici tedavilerinde büyük bir atılım gerçekleştirdi.
Yürütülmekte Olan Araştırma – Gen Düzenleme
CRISPR-Cas9** ve diğer gen düzenleme teknolojileri, talasemi için potansiyel iyileştirici tedaviler olarak araştırılmakta ve klinik deneyler umut verici erken sonuçlar göstermektedir.
Bu kilometre taşları, geçtiğimiz yüzyılda talaseminin teşhisi, tedavisi ve potansiyel tedavisinde kaydedilen önemli ilerlemeleri göstermektedir.
İleri Okuma
Olivieri, N. F., & Brittenham, G. M. (1997). Iron-chelating therapy and the treatment of thalassemia. Blood, 89(3), 739-761.
Cao, A., Galanello, R., & Rosatelli, M. C. (1997). Prenatal diagnosis and screening of the hemoglobinopathies. Baillière’s Clinical Haematology, 10(2), 203-222.
Rund, D., & Rachmilewitz, E. (2005). Beta-thalassemia. New England Journal of Medicine, 353(11), 1135-1146.
Vichinsky, E. P. (2005). Changing patterns of thalassemia worldwide. Annals of the New York Academy of Sciences, 1054(1), 18-24.
Modell, B., & Darlison, M. (2008). Global epidemiology of haemoglobin disorders and derived service indicators. Bulletin of the World Health Organization, 86, 480-487.
Cao, A., & Galanello, R. (2010). Beta-thalassemia. Genetics in Medicine, 12(2), 61-76.
Galanello, R., & Origa, R. (2010). Beta-thalassemia. Orphanet journal of rare diseases, 5(1), 1-12.
Galanello, R., & Origa, R. (2010). Alpha-thalassemia. Orphanet Journal of Rare Diseases, 5(1), 1-15.
Weatherall, D. J. (2010). The inherited diseases of hemoglobin are an emerging global health burden. Blood, 115(22), 4331-4336.
Borgna-Pignatti, C., & Gamberini, M. R. (2011). Complications of thalassemia major and their treatment. Expert Review of Hematology, 4(3), 353-366.
Cappellini, M. D., Cohen, A., Porter, J., Taher, A., & Viprakasit, V. (Eds.). (2014). Guidelines for the management of transfusion dependent thalassemia (TDT) (3rd ed.). Thalassemia International Federation.
Angelucci, E., Matthes-Martin, S., Baronciani, D., Bernaudin, F., Zecca, M., Maury, S., & Locatelli, F. (2014). Hematopoietic stem cell transplantation in thalassemia major. Haematologica, 99(5), 833-840.
Origa, R. (2017). Beta-Thalassemia. Genereviews.
Lal, A., & Goldrich, M. L. (2020). Alpha Thalassemia. In StatPearls [Internet]. StatPearls Publishing.
Taher, A. T., Musallam, K. M., & Cappellini, M. D. (2021). Thalassemia. The Lancet, 397(10291), 369-384.





















Yorum yazabilmek için oturum açmalısınız.