İçindekiler
Tanım ve Patofizyoloji
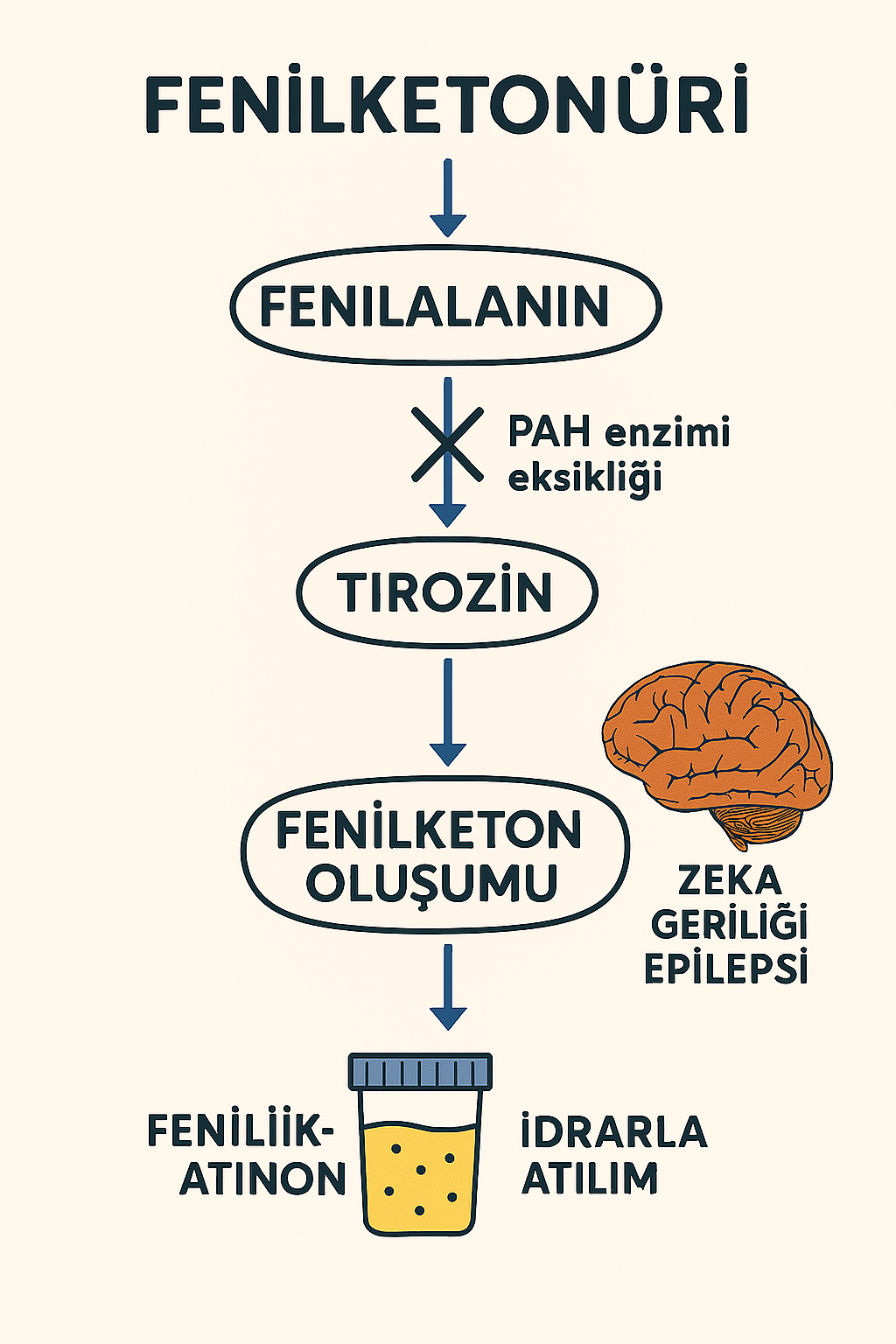
Fenilketonüri (PKU), amino asit metabolizmasında yer alan fenilalanin hidroksilaz (PAH) enzimindeki genetik defekt nedeniyle ortaya çıkan, otozomal resesif geçişli konjenital bir metabolik bozukluktur. Bu enzim fenilalanini, tirozin amino asidine hidroksile eder. PAH enzim aktivitesinin azalmış ya da yok olması sonucu, fenilalanin organizmada birikir ve başta santral sinir sistemi olmak üzere çok sayıda fizyolojik sistem üzerinde toksik etkiler oluşturur.
Patofizyolojik olarak, yüksek fenilalanin düzeyleri beyin gelişimini olumsuz etkiler; miyelin sentezini bozar, sinaptogenezde aksamalara yol açar ve nörotransmitter (dopamin, norepinefrin, serotonin) sentezini sekteye uğratır. Bu süreçler kümülatif olarak mental retardasyon, epileptik nöbetler, davranış bozuklukları ve motor beceri geriliği gibi klinik belirtilerle sonuçlanır.
Epidemiyoloji
PKU dünya genelinde nadir görülmekle birlikte, insidansı ülkeden ülkeye değişkenlik göstermektedir. Avrupa genelinde yaklaşık olarak her 7.000 canlı doğumda 1 görülürken, Türkiye’de bu oran yaklaşık 1:2.600 gibi yüksek bir düzeye ulaşmaktadır. Bu oran, yüksek taşıyıcılık sıklığı (%1.5-2) ve akraba evliliklerinin yaygınlığı ile ilişkilidir.
Genetik geçiş otozomal resesiftir. Yani hastalık, bireyin her iki ebeveyninden de mutant PAH geni alması durumunda ortaya çıkar. Taşıyıcı bireylerde klinik bulgu yoktur ancak hastalık nesilden nesile sessizce taşınabilir.
Klinik Bulgular
Tedavi edilmeyen PKU hastalarında:
- Zihinsel gelişim geriliği
- Nöbetler (epilepsi)
- Davranışsal sorunlar
- Egzama benzeri dermatit
- Açık ten, sarı saç, mavi göz gibi fenotipik değişiklikler (melanin sentezinde tirozin azalmasına bağlı)
- “Küf benzeri” idrar kokusu (fenilketon atılımına bağlı)
Fenilketonüri ismi, idrarda biriken fenilketon maddesinden kaynaklanmaktadır. Bu bileşik, fenilalanin metabolizmasının yıkım ürünlerinden biridir ve idrarla atılır.
Tanı
PKU, doğumdan sonraki ilk günlerde yapılan yenidoğan topuk kanı taraması (Guthrie testi, tandem kütle spektrometrisi) ile tespit edilebilir. Erken tanı, kalıcı beyin hasarını önlemek açısından kritik öneme sahiptir. Plazma fenilalanin düzeyinin > 6 mg/dL (360 µmol/L) olması tanısal öneme sahiptir.
Tedavi Yaklaşımları
1. Diyet Tedavisi
Fenilketonürinin temel tedavi yöntemi, fenilalanin içeriği düşük, protein kısıtlı diyet uygulanmasıdır. Fenilalanin doğada tüm proteinlerde bulunduğundan, hastalar için özel olarak formüle edilmiş aminoasit karışımları (fenilalanin içermeyen tıbbi mamalar) kullanılır.
- Diyet tedavisine yenidoğan döneminde başlanmalıdır.
- Amaç, plazma fenilalanin düzeyini 2-4 mg/dL (120-240 µmol/L) aralığında tutmaktır.
- Diyet tedavisi ideal olarak erişkinliğe dek, bazı kaynaklara göre ömür boyu sürdürülmelidir.
- Diyet ihlali, nörokognitif fonksiyonlarda bozulmalara, duygu durum değişikliklerine ve dikkat eksikliği gibi nöropsikiyatrik sorunlara yol açabilir.
2. Tetrahidrobiopterin (BH₄; Sapropterin) Tedavisi
Tetrahidrobiopterin (BH₄), PAH enziminin kofaktörüdür. Bazı hastalarda PAH enziminin aktivitesi BH₄ ile artırılabilir.
- Klasik PKU hastalarının yaklaşık %30–50’si BH₄ yanıtlıdır.
- BH₄ duyarlılığı, oral yükleme testi ile belirlenir.
- BH₄ aynı zamanda dopamin ve serotonin sentezinde de görev alır; eksikliğinde bu nörotransmitterlerin düzeyi azalabilir.
- Bu eksikliklerde L-Dopa ve 5-hidroksitriptofan ile ikame tedavisi uygulanır.
- Tedavi takibi için beyin omurilik sıvısında (BOS) homovanilik asit (dopamin yıkım ürünü) ve 5-HIAA düzeyleri izlenir.
3. Enzim Replasman Tedavisi
Pegvaliaz, 2019 yılında Avrupa İlaç Ajansı (EMA) tarafından onaylanan, PKU tedavisinde kullanılan bir enzim replasman tedavisidir.
- Fenilalanin amonyak liyaz (PAL) içeren pegilasyonlu bir enzimdir.
- Bu enzim, PAH’ın yerine geçerek fenilalanini doğrudan parçalar.
- Oral diyet kısıtlamasına alternatif ya da destekleyici bir seçenek olarak sunulur.
- Tedavi sırasında immünolojik yan etkiler (örn. antikor gelişimi, anaflaksi) izlenmelidir.
Keşif
Fenilketonüri’nin (PKU) keşif tarihi, genetik hastalıklar ve metabolizma bozuklukları tarihinde oldukça önemli bir dönüm noktasıdır. Aşağıda kronolojik olarak yapılandırılmış şekilde PKU’nun bilimsel keşfi ve bu alandaki önemli gelişmeler sunulmaktadır:
Fenilketonüri’nin Keşif Tarihi
1934 – Asbjørn Følling’in Keşfi
- Norveçli doktor ve kimyager Asbjørn Følling, 1934 yılında Oslo’da zihinsel engelli iki kardeş üzerinde yaptığı araştırmalar sırasında, bu çocukların idrarında fenilpirüvik asit adlı alışılmadık bir metabolitin yüksek miktarda bulunduğunu fark etti.
- Bu çocukların zihinsel engelinin, fenilalanin metabolizmasındaki bir bozukluk sonucu geliştiğini gösterdi.
- Følling, bu bozukluğu “oligophrenia phenylpyruvica” (fenilpirüvik oligofreni) olarak adlandırdı.
- Bu keşif, ilk kalıtsal metabolik hastalıklardan birinin biyokimyasal düzeyde tanımlanması anlamına gelir.
1940’lar – Genetik Temelin Fark Edilmesi
- 1940’lı yıllarda, PKU’nun otozomal resesif kalıtımla geçtiği anlaşılmaya başlandı.
- Bu dönemde, PKU’nun taşıyıcılık ve ebeveynlerin taşıyıcı olup çocukta hastalık gelişmesi gibi Mendel kalıtımı ilkeleriyle ilişkisi netleştirildi.
1953 – Horst Bickel ve Diyet Tedavisinin Başlatılması
- Alman doktor Horst Bickel, İngiltere’de zihinsel engelli bir PKU hastasına fenilalaninden yoksun bir diyet uyguladı.
- Bu uygulama sonucunda çocuğun klinik tablosunda belirgin düzelmeler görüldü.
- Bu gelişme, tıbbi diyetetik tedavinin klinik genetikte ilk başarılarından biri olarak kabul edilir.
1960’lar – Yenidoğan Tarama Programlarının Başlaması
- Dr. Robert Guthrie, 1961 yılında PKU’yu taramak üzere bakteriyel inhibitör testi (Guthrie testi) geliştirdi.
- Bu test ile fenilalanin yüksekliğine dayalı olarak yenidoğan topuk kanı taraması mümkün hale geldi.
- 1963’te ABD’de bazı eyaletlerde PKU taraması zorunlu hale geldi; ardından Avrupa ülkeleri ve Türkiye’de de uygulamaya konuldu.
1980’ler – Moleküler Genetik Düzeyde Tanımlama
- 1980’lerin sonlarına doğru PAH geninin yapısı tanımlandı.
- 1989 yılında, 12. kromozomdaki PAH geninde mutasyonların PKU’ya yol açtığı moleküler düzeyde gösterildi.
- Bu, hastalığın moleküler genetik tanısı ve prenatal tanı olanaklarını doğurdu.
2000’ler – Yeni Tedavi Yaklaşımları
- 2007: FDA, tetrahidrobiopterin (sapropterin dihidroklorür) tedavisini onayladı (BH4 yanıtlı PKU için).
- 2018–2019: Avrupa İlaç Ajansı (EMA), pegvaliaz adlı enzim replasman tedavisini onayladı.
- Bu tedaviler, klasik diyet tedavisine alternatif veya tamamlayıcı yeni yolların gelişimini simgelemektedir.
İleri Okuma
- Følling, A. (1934). Über Ausscheidung von Phenylbrenztraubensäure in den Harn als Stoffwechselanomalie in Verbindung mit Imbezillität. Hoppe-Seyler’s Zeitschrift für Physiologische Chemie, 227(3-4), 169–176.
- Bickel, H., Gerrard, J., & Hickmans, E. M. (1953). The influence of phenylalanine intake on the chemistry and behaviour of a phenylketonuric child. Acta Paediatrica, 42(1), 64–77.
- Guthrie, R., & Susi, A. (1963). A Simple Phenylalanine Method for Detecting Phenylketonuria in Large Populations of Newborn Infants. Pediatrics, 32(3), 338–343.
- DiLella, A. G., et al. (1986). Molecular structure and polymorphic map of the human phenylalanine hydroxylase gene. Biochemistry, 25(4), 743–749.
- Levy, H. L. (2007). Sapropterin hydrochloride: oral therapy for phenylketonuria. Expert Opinion on Investigational Drugs, 16(2), 239–251.
- Blau, N., van Spronsen, F. J., & Levy, H. L. (2010). Phenylketonuria. The Lancet, 376(9750), 1417–1427.
- van Wegberg, A. M. J., MacDonald, A., Ahring, K., et al. (2017). The complete European guidelines on phenylketonuria: diagnosis and treatment. Orphanet Journal of Rare Diseases, 12, Article 162.
- Longo, N., Arnold, G. L., et al. (2018). Pegvaliase for the treatment of phenylketonuria: A review of clinical studies. Drug Design, Development and Therapy, 12, 313–324.
- Longo, N., Singh, R. H., Harding, C. O., et al. (2019). International clinical guidelines for the treatment of phenylketonuria. Molecular Genetics and Metabolism, 127(1), 1–5.
- Singh, R. H., Cunningham, A. C., Mofidi, S., et al. (2020). Updated recommendations for the nutrition management of phenylalanine hydroxylase deficiency. Genetics in Medicine, 22(5), 1106–1117.
- Bik-Multanowski, M., Didycz, B., Mozrzymas, R., et al. (2023). Pegvaliase therapy in patients with phenylketonuria: A multicenter study from Central and Eastern Europe. Orphanet Journal of Rare Diseases, 18, Article 145.