
Tanım ve Genel Bakış
Trombotik Trombositopenik Purpura (TTP), nadir fakat potansiyel olarak yaşamı tehdit eden bir trombotik mikroanjiyopati alt tipidir. Hastalık, başta trombositopeni ve mikroanjiyopatik hemolitik anemi (MAHA) olmak üzere, çoklu organ tutulumu ile seyreden bir klinik tabloya neden olur. TTP’nin ayırt edici özelliği, küçük damar sisteminde (mikrovasküler yapılar) trombositlerin aşırı aktivasyonu ve agregasyonu sonucu gelişen mikrotrombus oluşumudur. Bu süreç, dolaşımda ultra büyük multimerik von Willebrand faktörü (UL-vWF) varlığında, ADAMTS13 enzim aktivitesinin ciddi oranda azalması ya da otoantikorlar ile inhibisyonu sonucu meydana gelir.
Patofizyoloji
TTP’nin temelinde, UL-vWF multimerlerinin proteolitik yıkımını gerçekleştiren bir metaloproteaz olan “A Disintegrin And Metalloprotease with Thrombospondin type 1 motif, member 13” (ADAMTS13) enziminin aktivitesinde belirgin bir azalma veya işlev kaybı yer alır. ADAMTS13 aktivitesi <%10 seviyelerine indiğinde, UL-vWF multimerleri dolaşımda birikerek trombositlerin spontan agregasyonunu indükler. Bu agregatlar, mikrovasküler yapılarda trombüs oluşumuna neden olurken, aynı zamanda eritrositlerin mekanik yıkımına yol açarak şistosit oluşumunu tetikler. Bu sürecin sonucunda:
- Trombositopeni: Trombositlerin mikrotrombus yapılarında tüketilmesi sonucu gelişir.
- Hemolitik Anemi: Mikrodamarlarda oluşan trombotik yapılar eritrositlerin parçalanmasına neden olur.
- Organ Hasarı: Beyin, böbrek ve kalp gibi yüksek perfüzyonlu organlarda iskemik hasarlar oluşabilir.
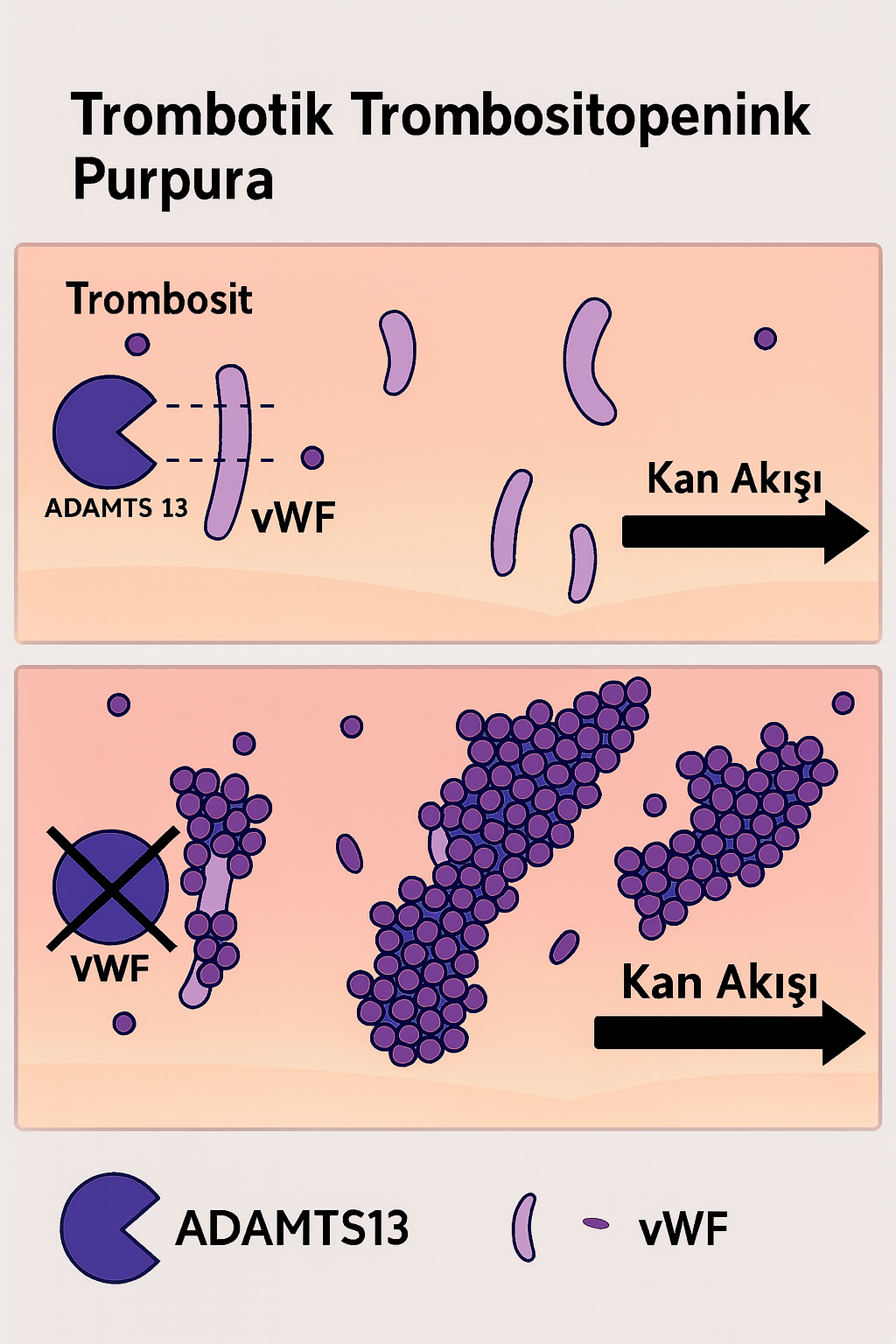
Etiyoloji
TTP, klinik olarak iki ana forma ayrılır:
1. Doğuştan (Konjenital) TTP (Upshaw-Schulman Sendromu)
- Nadir görülen otozomal resesif geçişli bir formdur.
- ADAMTS13 geninde meydana gelen bialelik mutasyonlar sonucu, enzimin miktarı ve/veya fonksiyonu yetersiz hale gelir.
- Genellikle çocukluk veya ergenlik döneminde başlar, enfeksiyon veya gebelik gibi tetikleyicilerle alevlenebilir.
2. Edinilmiş TTP
- En sık görülen formdur.
- ADAMTS13’e karşı gelişen otoimmün IgG antikorları enzimi inhibe eder.
- Kadınlarda daha sık görülür, sistemik lupus eritematozus (SLE), HIV, gebelik ve bazı ilaçlar (ör. ticlopidin, klopidogrel, kemoterapötikler) ile ilişkilendirilmiştir.
Klinik Belirti ve Bulgular
Klasik olarak “pentad” olarak adlandırılan tablo her hastada görülmeyebilir. Günümüzde TTP tanısı için trombositopeni ve MAHA varlığı çoğu zaman yeterlidir. Klinik özellikler şunlardır:
- Trombositopeni: Peteşi, purpura, kolay morarma, burun kanaması gibi kanama belirtileri.
- Mikroanjiyopatik Hemolitik Anemi (MAHA): Şistosit varlığı, yorgunluk, solukluk, taşikardi.
- Nörolojik Bulgular: Baş ağrısı, konfüzyon, geçici iskemik atak, nöbetler, koma.
- Renal Disfonksiyon: Hematüri, proteinüri, artmış serum kreatinini (TTP’de tipik olarak HÜS’ten daha hafiftir).
- Ateş: Klasik tanımda yer alsa da günümüzde nadir görülür ve özgül değildir.
Tanısal Yaklaşım
TTP tanısı acil bir durum olup, klinik şüpheyle birlikte tanısal süreç başlatılmalı ve tedavi gecikmeden uygulanmalıdır. Aşağıdaki testler tanı koymada yardımcıdır:
Laboratuvar Bulguları:
| Parametre | Bulgular |
|---|---|
| Trombosit sayısı | Düşük (<30.000/μL sıklıkla) |
| Hemoglobin | Düşük |
| LDH (Laktat Dehidrogenaz) | Yüksek – hemoliz ve doku hasarı göstergesi |
| Dolaylı Bilirubin | Artmış |
| Haptoglobin | Düşük (serbest hemoglobine bağlandığı için tükenir) |
| Retikülosit | Artmış |
| Periferik Yayma | Şistositler (fragmentositoz) |
| ADAMTS13 aktivite düzeyi | <%10 → tanı için yüksek özgüllük |
| ADAMTS13 inhibitörleri | Edinilmiş formlarda pozitif |
Ayırıcı Tanılar:
- HÜS (Hemolitik Üremik Sendrom): Özellikle çocuklarda; TTP’ye göre böbrek tutulumu daha belirgindir.
- DIC (Yaygın İntravasküler Koagülasyon): PT/aPTT uzaması ve D-dimer artışı tipiktir.
- HELLP Sendromu: Gebelikle ilişkili; hemoliz, karaciğer enzim yüksekliği, trombositopeni üçlüsüyle seyreder.
Tedavi Yaklaşımı
1. Plazmaferez (PEX – Plazma değişim tedavisi):
- TTP tedavisinin temelini oluşturur.
- Günde bir kez uygulanan tedavi ile hem inhibitör antikorlar temizlenir hem de ADAMTS13 replasmanı sağlanır.
- Tedavi, klinik düzelme ve laboratuvar parametrelerinin normale dönmesine kadar sürdürülür.
2. İmmünosupresyon:
- Edinilmiş TTP’de otoantikor üretiminin baskılanması hedeflenir.
- Kortikosteroidler (örn. prednizon 1 mg/kg/gün) ilk basamak tedavidir.
- Dirençli veya tekrarlayan olgularda:
- Rituksimab: Anti-CD20 monoklonal antikoru; B hücrelerini hedef alır.
- Siklofosfamid veya diğer immünsüpresif ajanlar da kullanılabilir.
3. Kaplacizumab:
- Anti-vWF nanomonomerik antikordur.
- vWF-trombosit etkileşimini bloke ederek mikrotrombus oluşumunu engeller.
- Klinik yanıt süresini kısaltır ve nüksü azaltır.
- EMA ve FDA onayı almıştır (2018, 2019).
4. Destekleyici Tedavi:
- RBC transfüzyonları: Semptomatik anemi varlığında.
- Trombosit transfüzyonu: Sadece hayatı tehdit eden kanamalarda (kanama riskini artırabilir).
- Organ fonksiyon takibi: Nefrolojik ve nörolojik izlem.
Prognoz ve Takip
- Uygun tedavi ile akut mortalite oranı <%10’a düşmüştür.
- Tedavi edilmeyen TTP’de ölüm oranı >%90 olarak bildirilmiştir.
- Edinilmiş formlarda nüks oranı %20–50 arasında değişebilir.
- Nüks riskine karşı ADAMTS13 düzeyleri ve inhibitör varlığı takiple izlenmelidir.
- Gebelik ve enfeksiyon gibi stresörler nüksü tetikleyebilir.
Keşif
İlk Olgu Bildirimi – 1924
- Eli Moschcowitz, 1924 yılında New York Mount Sinai Hospital’da 16 yaşındaki bir kız hastada görülen ani başlangıçlı hemolitik anemi, peteşi, nörolojik bozulma ve otopsi sonrası mikrovasküler trombüs bulgularını tanımlamıştır.
- Moschcowitz bu tabloyu “akut afibrinojenemik purpura” olarak adlandırmıştır. Ancak aslında tanımladığı klinik tablo, günümüzde TTP olarak adlandırdığımız hastalıktır.
2. Klinik Sendromun Netleşmesi – 1960’lar
- 1960’lı yıllarda, TTP artık klasik “pentad” olarak bilinen semptomlar bütünüyle tanımlanmaya başlanmıştır:
- Trombositopeni
- Mikroanjiyopatik hemolitik anemi
- Nörolojik bulgular
- Ateş
- Renal disfonksiyon
- Bu yıllarda, hastalığın farklı klinik formları ve atipik seyirli vakaları da tanımlanmıştır.
3. ADAMTS13’ün Keşfi – 2001
- 1990’ların sonlarından itibaren, hastalığın altında yatan moleküler mekanizmanın von Willebrand faktörü (vWF) ve onun düzenleyici enzimleriyle ilişkili olduğu fark edilmiştir.
- 2001 yılında, bağımsız araştırma ekipleri tarafından ADAMTS13 enzimi tanımlanmış ve TTP ile ilişkisi net olarak ortaya konmuştur:
- ADAMTS13’ün eksikliği veya inhibitör otoantikorlar tarafından etkisiz hale getirilmesi, UL-vWF multimerlerinin yıkılamamasına ve dolayısıyla mikrotrombus oluşumuna neden olmaktadır.
4. Edinilmiş ve Konjenital TTP’nin Ayrımı – 2000’li Yıllar
- ADAMTS13 testlerinin klinik pratikte yaygınlaşmasıyla birlikte, TTP’nin konjenital (Upshaw-Schulman sendromu) ve edinilmiş (otoimmün) formları net olarak ayrılmıştır.
- Edinilmiş TTP’de ADAMTS13’e karşı nötralizan otoantikorlar, konjenital TTP’de ise genetik mutasyonlar temel neden olarak belirlenmiştir.
5. Kaplacizumab ve Hedefe Yönelik Tedavi – 2017–2019
- 2017’de faz II HERCULES çalışmasıyla birlikte kaplacizumab, UL-vWF’ye karşı geliştirilen ilk hedefe yönelik biyolojik tedavi olarak tanıtılmıştır.
- 2018 yılında Avrupa İlaç Ajansı (EMA) ve 2019 yılında Amerikan Gıda ve İlaç Dairesi (FDA) tarafından onay almıştır.
İleri Okuma
- Tsai, H. M. (2003). Pathophysiology of thrombotic thrombocytopenic purpura. International Journal of Hematology, 77(1), 1–19.
- George, J. N., & Nester, C. M. (2014). Syndromes of thrombotic microangiopathy. New England Journal of Medicine, 371(7), 654–666.
- Scully, M. et al. (2019). Caplacizumab treatment for acquired thrombotic thrombocytopenic purpura. New England Journal of Medicine, 380(4), 335–346.
- Zheng, X. L. et al. (2020). International Working Group consensus: Diagnosis and treatment of TTP. Blood Advances, 4(23), 6250–6273.
- Sadler, J. E. (2021). Von Willebrand factor, ADAMTS13, and thrombotic thrombocytopenic purpura. Blood, 138(15), 1150–1160.
- Furlan, M., Robles, R., & Lämmle, B. (1998). “Partial purification and characterization of a protease from human plasma cleaving von Willebrand factor to fragments produced by in vivo proteolysis.” Blood, 91(12), 4736–4743.
- Tsai, H. M., & Lian, E. C. Y. (1998). “Antibodies to von Willebrand factor–cleaving protease in acute thrombotic thrombocytopenic purpura.” New England Journal of Medicine, 339(22), 1585–1594.
- Lämmle, B., Kremer Hovinga, J. A., & George, J. N. (2005). “Thrombotic thrombocytopenic purpura.” Lancet, 366(9482), 1487–1499.
- Scully, M., et al. (2019). “Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies.” British Journal of Haematology, 185(3), 460–478.
Yorum yazabilmek için oturum açmalısınız.