Kromozomal sapmalar, insan hücrelerindeki kromozomların tipik sayısından (sayısal sapmalar) veya yapısından (yapısal sapmalar) sapmaları ifade eder.
Kromozomal Aberasyonların Epidemiyolojisi
Kromozomal anormalliklerin yaklaşık olarak her 200 doğumdan 1’inde meydana geldiği tahmin edilmektedir, ancak bu istatistik yalnızca yaşamla uyumlu olan anormallikleri hesaba katmaktadır. Çok daha fazla kromozomal anormallik rahimde meydana gelir ve erken gebelik kayıplarından sorumludur. Aslında, ilk üç aylık dönemdeki düşüklerin** %50’sinin kromozomal anormalliklerden kaynaklandığı tahmin edilmektedir. Kromozomal sapmaların sıklığı, mayoz bölünme sırasındaki hatalar nedeniyle ebeveyn yaşı, özellikle de anne yaşı** ile artma eğilimindedir.

Kromozomal Aberasyonların Sınıflandırılması

Kromozomal sapmaların iki temel şekli vardır:
1. Sayısal Kromozomal Aberasyonlar
Bunlar, normal diploid kromozom sayısından (insanlarda 46) sapmaları içerir.
- Poliploidi: Fazladan bir kromozom setinin tamamının varlığı. Poliploidi (örneğin, 69 kromozomun bulunduğu triploidi) tipik olarak insan yaşamıyla bağdaşmaz ve genellikle erken embriyonik ölüme yol açar.
- Neuploidi**: Haploid setin (insanlarda 23) bir katı olmayan anormal sayıda kromozomun varlığı. Genellikle mayoz bölünme sırasında homolog kromozomların veya kardeş kromatidlerin düzgün şekilde ayrılamadığı *nondisjunction* durumundan kaynaklanır. Bu durum fazladan bir kromozoma (trizomi, 2n + 1) veya eksik bir kromozoma (monozomi, 2n – 1) sahip hücrelerle sonuçlanabilir.
- Trizomi**: İnsanlarda en sık görülen canlı trizomiler şunlardır:
- Trizomi 21 (Down sendromu)**: Kromozom 21’in fazladan bir kopyasının varlığı. Zihinsel engelliliğin en yaygın kromozomal nedenidir.
- Trisomi 13 (Pätau sendromu): Ciddi zihinsel engellilik ve çoklu konjenital anormalliklerle karakterizedir. Etkilenen bireylerin çoğu yaşamın ilk yılından sonra hayatta kalamaz.
- Trisomi 18 (Edwards sendromu): Yine ciddi zihinsel ve fiziksel engellerle ilişkilidir ve yaşamın ilk yılında yüksek ölüm oranı vardır.
- Monozomi**: Otozomal monozomilerin çoğu yaşamla bağdaşmaz ve erken düşüklere yol açar. Bununla birlikte, bir bireyin tek bir X kromozomuna (45,X) sahip olduğu *monozomi X (Turner sendromu)* yaşamla uyumludur, ancak fiziksel anormallikler, kısırlık ve kardiyovasküler sorunlarla ilişkilidir.
- Mozaiklik**: Bu durum, aynı bireyde farklı kromozomal yapılara sahip iki veya daha fazla hücre popülasyonu bulunduğunda ortaya çıkar. Mozaiklik döllenmeden sonra mitoz bölünmedeki hatalardan kaynaklanabilir ve etkilenen hücrelerin oranına bağlı olarak kromozomal anormalliklerin daha hafif veya daha değişken fenotipik ifadesine yol açabilir.
- Trizomi**: İnsanlarda en sık görülen canlı trizomiler şunlardır:
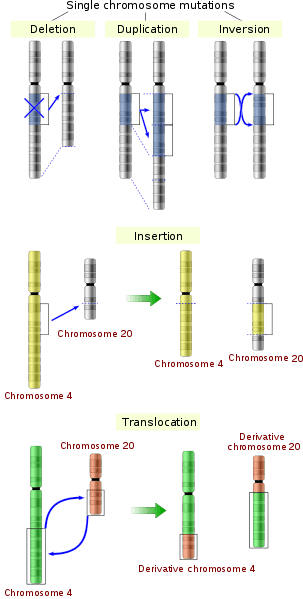
2. Yapısal Kromozomal Aberasyonlar
Yapısal sapmalar, kromozomlardaki kırılmalar ve parçaların uygunsuz şekilde yeniden birleşmesi nedeniyle meydana gelir. Bunlar, kırılma noktalarından etkilenen genlere bağlı olarak önemli klinik etkilere sahip olabilir.
- Translokasyonlar: Bir kromozomun bir segmenti başka bir kromozoma aktarıldığında meydana gelir. Bu rekiprokal (iki kromozom arasında değiş tokuş edilen segmentler) ya da Robertsonian (iki akrosentrik kromozomun uzun kollarının füzyonu) olabilir. Örneğin, 21. kromozomu içeren Robertsonian translokasyonlar herediter Down sendromu ile sonuçlanabilir.
- Silinmeler**: Bir kromozom segmentinin kaybı. En iyi bilinen örneklerden biri, kromozom 5’in (5p) kısa kolundaki bir delesyonun neden olduğu *cri du chat sendromudur*. Bu durum zihinsel engellilik ve kedininkine benzeyen tiz bir ağlama ile karakterizedir.
- İnversiyonlar**: Bir kromozom segmenti tersine döndüğünde meydana gelir. İnversiyonlar *parasentrik* (sentromeri içermeyen) veya perisentrik (sentromeri içeren) olabilir. Mayoz bölünme sırasında komplikasyonlara yol açabilir ve dengesiz gametlere neden olabilirler.
- İzokromozomlar**: Bir kromozom anormal bir düzlem boyunca bölünerek iki özdeş kola yol açtığında oluşur. Bu durum genetik dengesizliklere yol açabilir.
- Halka Kromozomlar**: Bir kromozomun uçları bir kırılmadan sonra birleştiğinde meydana gelir. Halka kromozomlar önemli gelişimsel ve tıbbi sorunlara neden olabilir.
Kromozomal Aberasyonların Teşhisi
Kromozomal sapmalar genellikle karyogramın üretildiği sitogenetik analiz yoluyla tespit edilir. Bir karyogram, bir bireyin kromozomlarının çiftler halinde düzenlenmiş bir fotoğrafıdır ve sayısal ve büyük yapısal anormalliklerin tanımlanmasına olanak tanır. Daha ince veya daha küçük sapmalar için floresan in situ hibridizasyon (FISH) gibi teknikler gerekebilir. FISH, spesifik kromozomal bölgeleri bağlamak için floresan problar kullanır ve mikrodelesyonların ve diğer küçük anormalliklerin tespit edilmesini sağlar.
- Doğum Öncesi Tanı**: Kromozom analizi, elde edilen fetal hücreler üzerinde gerçekleştirilebilir:
- Amniyosentez (gebeliğin 15. haftasından itibaren)
- Koryon villus örneklemesi (gebeliğin 11. haftasından itibaren)
Bu prosedürler gelişmekte olan fetüsteki anöploidileri ve bazı yapısal anormallikleri tespit edebilir.
Kombine Tarama Testleri
Kombine prenatal tarama testleri, kromozomal anormalliklerin, özellikle de trizomilerin erken belirtilerini sağlayabilir. Bu testler şunları içerir:
- Fetal ense kalınlığı (NT): Fetal boynun arka kısmındaki sıvı dolu boşluğun ultrasonla ölçümü. Artmış ense kalınlığı Down sendromu gibi kromozomal anormalliklerle ilişkilidir.
- Anne serum belirteçleri**:
- Gebelikle ilişkili plazma proteini-A (PAPP-A)**: Düşük seviyeler daha yüksek kromozomal anormallik riski ile ilişkilidir.
- Serbest beta-hCG**: Yüksek seviyeler trizomi 21’in göstergesi olabilir.
- Hücresiz fetal DNA (cfDNA)**: Non-invaziv prenatal test (NIPT), trizomi 21, 13 ve 18 dahil olmak üzere kromozomal anormallikleri tespit etmek için annenin kanındaki fetal DNA parçalarını analiz eder.

Kromozomal Aberasyonlar ve Kanser
Yapısal kromozomal anormallikler genellikle kanserlerle, özellikle de lösemi ile ilişkilidir. Örneğin, kromozom 9 ve 22 arasında bir translokasyon olan Philadelphia kromozomu kronik miyelojenöz löseminin (KML) ayırt edici özelliğidir. Somatik hücrelerdeki kromozomal delesyonlar, duplikasyonlar veya translokasyonlar, hücre büyümesini düzenleyen genleri bozarak kontrolsüz lösemiye yol açabilir.
Keşif
1. Kromozomların Keşfi (1888)
- İlk olarak Walther Flemming hücre çekirdeğinde kromatini gözlemledi ve bu da kromozomların keşfine yol açtı. Daha sonra Heinrich Wilhelm Waldeyer 1888 yılında “kromozom ” terimini ortaya attı. Bu, genetik materyali ve kromozomal anormallikleri anlamanın temelini attı.
2. Kromozomal Kalıtım Teorisi (1902)
- Walter Sutton** ve Theodor Boveri bağımsız olarak kromozomal kalıtım teorisini formüle ederek kromozomların genetik bilgi taşıdığını öne sürmüşlerdir. Bu teori, kromozomal anormallikleri genetik bozukluklara bağlamak için çok önemliydi.
3. Tanımlanan İlk Sayısal Kromozomal Aberasyon (1959)
- Jérôme Lejeune**, *Down sendromunun* 21. kromozomun fazladan bir kopyasından (trisomi 21) kaynaklandığını keşfetti. Bu, ilk kez bir insan genetik bozukluğunun kromozomal bir anormallikle ilişkilendirilmesiydi.
4. Karyotipleme Gelişimi (1956)
- Joe Hin Tjio** ve Albert Levan insanların 46 kromozoma (23 çift) sahip olduğunu belirledi. Bu, trizomiler ve monozomiler gibi sayısal kromozomal sapmaların teşhisinde önemli bir adımdı.
5. İlk Yapısal Kromozomal Anormallik (1960)
- Kromozom 9 ve 22 arasında bir karşılıklı translokasyon olan Philadelphia kromozomu, Peter Nowell ve David Hungerford tarafından keşfedilmiştir. Kanserle (kronik miyeloid lösemi) bağlantılı ilk yapısal kromozomal anormalliktir.
6. Amniyosentez Yoluyla Doğum Öncesi Teşhis (1966)
- Kromozomal analiz için fetal hücrelerin elde edilmesinde amniyosentezin kullanılması, trizomi 21 gibi kromozomal anormalliklerin doğum öncesi teşhisine olanak sağlamıştır. Bu teknik doğum öncesi taramada devrim yarattı.
7. Floresan İn Situ Hibridizasyon (FISH) Gelişimi (1980’ler)
- FISH**, kromozomlar üzerindeki spesifik DNA dizilerini tespit etmek için floresan problar kullanan bir sitogenetik teknik olarak geliştirilmiştir. Kromozomal aberasyonlarda mikrodelesyonları ve translokasyonları tespit etmek için güçlü bir araç haline geldi.
8. Turner Sendromunun Moleküler Anlayışı (1990’lar)
- Turner sendromu (45,X)** üzerine yapılan araştırmalar, cinsiyet kromozomlarındaki monozomilerin gelişimi nasıl etkilediğine dair içgörüler sağlamış ve dozaj dengesizliklerinin gen ifadesini nasıl etkilediğini ortaya koymuştur.
9. Hücresiz Fetal DNA Testinin (cfDNA) Tanıtımı (2011)
- Anne kanında cfDNA kullanılarak yapılan non-invaziv prenatal testler, amniyosentez gibi invaziv prosedürler olmaksızın 21, 13 ve 18 gibi trizomilerin erken tespit edilmesini sağlayarak fetüs üzerindeki riski azaltmıştır.
10. İnsan Genom Projesi (2003)
- İnsan Genom Projesi’nin** tamamlanması, insan genomunun tam bir haritasının çıkarılmasını sağlayarak kromozomal yapısal anormalliklerin genetik temelinin ve hastalıklardaki rollerinin daha iyi anlaşılmasını kolaylaştırdı.
11. CRISPR-Cas9 Genom Düzenleme (2012)
- CRISPR-Cas9** teknolojisinin keşfi, kromozomal anormalliklerin genetik düzeyde düzeltilmesine yönelik potansiyel terapötik yaklaşımlara kapı açarak kromozomal bozuklukların gelecekteki tedavileri için umut verdi.
İleri Okuma
- Flemming, W. (1882). “Zellsubstanz, Kern und Zelltheilung.” Leipzig: Verlag von FCW Vogel.
- Sutton, W. S. (1902). “On the morphology of the chromosome group in Brachystola magna.” Biological Bulletin, 4(1), 24-39.
- Lejeune, J., Gautier, M., & Turpin, R. (1959). “Study of somatic chromosomes from 9 mongoloid children.” Comptes Rendus Hebdomadaires des Séances de l’Académie des Sciences, 248, 1721-1722.
- Nowell, P. C., & Hungerford, D. A. (1960). “A minute chromosome in human chronic granulocytic leukemia.” Science, 132(3438), 1497-1499.
- Zaloudek, C., & Worsham, M. J. (1991). “FISH techniques and their application in medical genetics.” Human Genetics, 86(1), 22-28
- Verma, R. S., & Babu, A. (1995). Human Chromosomes: Principles and Techniques (2nd ed.). McGraw-Hill.
- Savage, J. R. K. (2000). “Classification and relationships of induced chromosomal structural changes.” Journal of Medical Genetics, 37(3), 185-189.
- Shaffer, L. G., & Lupski, J. R. (2000). “Molecular mechanisms for constitutional chromosomal rearrangements in humans.” Annual Review of Genetics, 34(1), 297-329.
- Hassold, T., & Hunt, P. (2001). “To err (meiotically) is human: The genesis of human aneuploidy.” Nature Reviews Genetics, 2(4), 280-291.
- Gardiner, K., Fortna, A., Bechtel, L., & Davisson, M. (2003). “Mouse models of Down syndrome: How useful can they be?” Trends in Genetics, 19(2), 79-85.
- Hochstenbach, R., Meijer, J. W., van Binsbergen, E., van Buuren, N., Ploos van Amstel, H. K., & Schuring-Blom, G. H. (2005). “Rapid detection of chromosomal aneuploidies in uncultured amniocytes by multiplex ligation-dependent probe amplification (MLPA).” Prenatal Diagnosis, 25(10), 998-1005.
- Nussbaum, R. L., McInnes, R. R., & Willard, H. F. (2007). Thompson & Thompson Genetics in Medicine (7th ed.). Saunders Elsevier.
- Pfundt, R., & Veltman, J. A. (2012). “Structural genomic variation in intellectual disability and related neurodevelopmental disorders.” Journal of Medical Genetics, 49(4), 217-227.
- Antonarakis, S. E. (2017). “Down syndrome and the complexity of genome dosage imbalance.” Nature Reviews Genetics, 18(3), 147-163.