
Epidemiyoloji
LCH nadir görülen bir hastalıktır ve görülme sıklığı 15 yaşın altındaki her bir milyon çocukta yaklaşık 4-8’dir. Çoğunlukla çocuklarda, özellikle de 1 ila 3 yaş arasında görülür. Daha nadir olmakla birlikte, yetişkinlerde de vakalar bildirilmiştir. Hastalık, yaklaşık 1,5:1 erkek-kadın oranı ile hafif bir erkek tercihi gösterir.
Etiyoloji ve Patofizyoloji
LCH’nin, kemik iliği kaynaklı olan ve mononükleer fagosit sisteminin (MPS) doku makrofajlarıyla özellikleri paylaşan Langerhans hücrelerinin klonal proliferasyonundan kaynaklandığı düşünülmektedir. Kesin nedeni belirsizliğini korusa da, BRAF genindeki mutasyonlar (özellikle BRAF V600E mutasyonu) gibi genetik mutasyonlar, LCH vakalarının yaklaşık %40-60’ında tespit edilmektedir. Bu mutasyonun mitojenle aktive olan protein kinaz (MAPK) yolunu aktive ederek kontrolsüz hücre büyümesine ve çoğalmasına katkıda bulunduğu bilinmektedir.
İçindekiler
Hücresel Özellikler
- İmmünohistokimyasal Belirteçler: LCH hücreleri aşağıdaki belirteçleri ifade eder:
- S-100: Tipik olarak Langerhans hücreleri gibi nöral krest kökenli hücrelerde bulunan bir protein belirteci.
- Vimentin: Genellikle mezenkimal kökenli hücrelerde ifade edilen bir tür ara filament proteini.
- CD1a: Langerhans hücrelerinin yüzeyinde ifade edilen bir glikoprotein.
- Birbeck Granülleri: Bunlar, Langerhans hücrelerinin karakteristik bir özelliği olarak elektron mikroskobu altında görülebilen pentalaminar, tenis raketi şeklindeki granüllerdir.
Bu moleküler ve histopatolojik belirteçler LCH’nin diğer histiyositoz türlerinden ayırt edilmesine yardımcı olur.
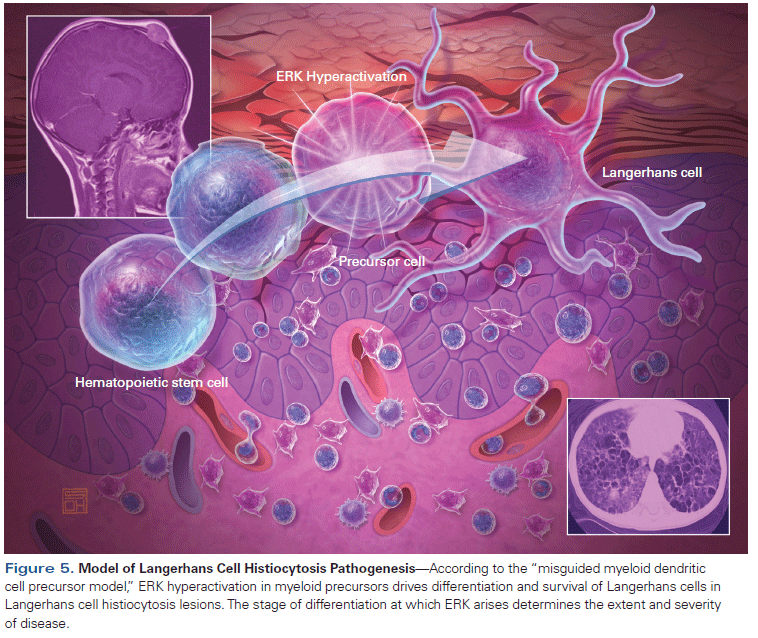
Klinik Sunum
LCH, klinik sunum açısından önemli ölçüde değişkenlik gösterebilir ve genellikle birbiriyle birleşen üç ana klinik formu vardır.
1. Abt-Letterer-Siwe Sendromu
- Yaş**: Tipik olarak bebeklerde yaşamın ilk yılı içinde ortaya çıkar.
- Semptomlar**: Nekrotize olabilen ve ikincil olarak enfekte olabilen sarı-kahverengi, pullu papüller. Hastalık genelleşir ve yüksek ateş, lenfadenopati, hepatosplenomegali, kemik değişiklikleri ve septisemiye yol açar. Bu formun seyri agresif ve genellikle ölümcüldür.
2. El-Schüller-Hıristiyan Hastalığı
- Yaş: Öncelikle çocukları etkiler.
- Semptomlar**: Özellikle baş, gövde ve anogenital bölgede kahverengi-kırmızı deri lezyonları. Bu lezyonlar kabuklanabilir, ülserleşebilir ve süperenfekte olabilir. Kemik lezyonları yaygındır ve diabetes insipidus (hipofiz bezi tutulumundan), ekzoftalmi (orbital granülomlardan dolayı) ve diğer sistemik belirtilere yol açar. Bu form ilerleyicidir ve genellikle ölümcüldür.
3. Eozinofilik Granülom
- Yaş**: Ağırlıklı olarak büyük çocukları ve genç yetişkinleri etkiler.
- Semptomlar**: Bu, öncelikle kemikleri etkileyen ve spontan kırıklara yol açan daha iyi huylu bir varyanttır. Hand-Schüller-Christian hastalığına benzer cilt değişiklikleri de olabilir. Bu form nispeten olumlu bir prognoza sahiptir ve kendiliğinden iyileşme mümkündür.

Teşhis Yaklaşımı
Klinik ve Görüntüleme Özellikleri
- Kemik Lezyonları**: LCH sıklıkla, özellikle kafatası, pelvis, kaburgalar ve uzun kemiklerde osteolitik lezyonlarla kendini gösterir. Osteoliz, röntgen veya bilgisayarlı tomografi (BT) taramalarında belirgin olabilir.
- “Cheerio İşareti ”**: LCH’nin akciğer tutulumunda görülür, merkezi bir kavitasyon alanı olan küçük nodüler lezyonlarla karakterizedir.
- Vertebra Plana: Bir omur gövdesinin düzleşmesi, LCH tanısı koydurabilir.
Biyopsi ve Histopatoloji
- Biyopsi: LCH tanısı tipik olarak biyopsi ile doğrulanır. Histopatolojik inceleme, yukarıda belirtilen immünohistokimyasal belirteçlerle Langerhans hücrelerinin proliferasyonunu gösterir.
- Ayırıcı Tanı**: LCH, Langerhans hücreli olmayan histiyositoz (örn. Erdheim-Chester hastalığı), seboreik dermatit ve Darier hastalığı dahil olmak üzere diğer histiyositik hastalıklardan ayırt edilmelidir.


Tedavi
Yerel Tedaviler
- Antiseptikler ve Glukokortikoidler**: Bunlar deri lezyonlarında iltihabı azaltmak ve ikincil enfeksiyonu önlemek için topikal olarak kullanılır.
Sistemik Tedaviler
- Kortikosteroidler**: Sistemik kortikosteroidler multisistem hastalığı için tedavinin temel taşıdır.
- Sitotoksik Ajanlar**: Metotreksat (MTX) ve 6-merkaptopürin, Langerhans hücre proliferasyonunu azaltmak için ciddi vakalarda kullanılır.
- İnterferon-alfa: Bu immünomodülatör bazı refrakter vakalarda kullanılabilir.
Radyasyon Tedavisi
- Öncelikli olarak kemik lezyonlarında, özellikle vertebra tutulumu veya şiddetli ağrı olan vakalarda endikedir.
Cerrahi Müdahaleler
Lokalize lezyonların cerrahi eksizyonu, özellikle kemik tutulumunda, zaman zaman uygulanmaktadır. Ancak radyasyon tedavisi daha çok inoperabl lezyonlar için kullanılır.
Prognoz
LCH’nin prognozu organ tutulumunun derecesine ve tanı yaşına bağlı olarak değişir. İzole, tek sistemli hastalıkta (örn. eozinofilik granülom), prognoz genellikle olumludur ve spontan rezolüsyon olasılığı vardır. Buna karşılık, özellikle karaciğer, akciğerler veya hematopoetik sistem gibi hayati organların tutulduğu çoklu sistem hastalığı daha kötü bir prognoz taşır. Abt-Letterer-Siwe sendromlu bebekler, agresif tedaviye rağmen genellikle ölümcül sonuçlara sahiptir.
Keşif
1. 1893: Histiyositik Hastalıkların Keşfi
- Paul Langerhans** ilk olarak insan derisinde daha sonra Langerhans hücreleri olarak bilinen dendritik hücreleri tanımlamıştır. Bu hücreler daha sonra LCH ile ilişkilendirilecektir.
2. 1921: Abt-Letterer-Siwe Hastalığı
- Erich Letterer** ve Siegfried Siwe** bebekleri etkileyen ciddi bir sistemik histiyositoz formunu tanımlamışlardır. Çoklu organ tutulumu ve ölümcül bir seyir ile karakterize olan bu form başlangıçta Letterer-Siwe hastalığı olarak adlandırılmıştır.
3. 1926: Hand-Schüller-Christian Hastalığı
- Alfred Hand, Arthur Schüller** ve Henry Christian diabetes insipidus, kemik lezyonları ve ekzoftalmi içeren bir LCH varyantı tanımlamıştır. Bu, çocuklarda LCH’nin farklı bir klinik sunumu olarak kabul edilmiştir.
4. 1940’lar: “Histiyositoz X “ Teriminin Ortaya Çıkışı
- Lichtenstein**, ortak bir histiyositik kökeni paylaşan bir hastalık spektrumunu (Abt-Letterer-Siwe hastalığı, Hand-Schüller-Christian hastalığı ve eozinofilik granülom) tanımlamak için “Histiositoz X” terimini ortaya atmıştır. İlgili histiyositlerin belirsiz doğasını vurgulamıştır.
5. 1960’lar: Elektron Mikroskopisi ve Birbeck Granülleri
- Elektron mikroskobu** Langerhans hücreleri içinde Birbeck granüllerini (tenis raketi şeklindeki organeller) tanımlayarak hastalık sürecindeki rollerini sağlamlaştırdı. Bu granüller önemli bir tanısal belirteç haline gelmiştir.
6. 1987: Langerhans Hücreli Histiyositoz olarak yeniden sınıflandırma
- Histiosit Topluluğu**, Langerhans hücrelerinin patogenezdeki rolünü vurgulayarak ve genel “Histiositoz X” teriminden uzaklaşarak hastalığı “Langerhans Hücreli Histiositoz” olarak yeniden sınıflandırdı. Bu yeniden sınıflandırma, ilgili spesifik hücresel patolojiyi vurgulamıştır.
7. 1990’lar: İmmünohistokimya ve Moleküler Belirteçler
- İmmünohistokimya** LCH’deki Langerhans hücrelerinin CD1a, S-100 ve langerin (CD207) eksprese ettiğini doğrulamıştır. Bu belirteçler, LCH’nin teşhisi için ayrılmaz hale gelmiş ve diğer histiyositozlardan ayırt edilmesine yardımcı olmuştur.
8. 2010: BRAF V600E Mutasyonunun Tanımlanması
- LCH vakalarının önemli bir kısmında BRAF V600E mutasyonlarının keşfedilmesi, hastalığın genetik temelinin anlaşılmasında bir dönüm noktası olmuştur. Bu mutasyon, MAPK yolunun aktivasyonu yoluyla kontrolsüz hücre çoğalmasına yol açmakta ve hedefe yönelik tedaviler için yollar açmaktadır.
9. 2012-2014: Hedefe Yönelik Tedavilerin Kullanımı
- BRAF V600E mutasyonu taşıyan LCH hastaları için BRAF inhibitörleri (örn. vemurafenib) gibi hedefli tedaviler kullanıma sunulmuştur. Bu, geleneksel kemoterapiden kişiselleştirilmiş tıbba önemli bir geçişi temsil etmektedir.
10. 2014: Dünya Sağlık Örgütü Tarafından Güncellenmiş Sınıflandırma
- Dünya Sağlık Örgütü (WHO)**, klonal kökenini ve onkojenik mutasyonların rolünü kabul ederek LCH’yi tamamen bir bağışıklık bozukluğundan ziyade neoplastik bir hastalık olarak sınıflandırmıştır.
11. 2015-Günümüz: Klinik Çalışmalar ve Araştırmalardaki Gelişmeler
- Son klinik çalışmalar, özellikle MAPK yolunu inhibe etmeyi amaçlayan hedefe yönelik tedavilere odaklanmıştır (örneğin, BRAF-negatif LCH vakalarında MEK inhibitörleri).
- Hayatta kalanlar için, özellikle nörodejeneratif hastalık ve ikincil maligniteler gibi komplikasyonların yönetiminde uzun vadeli sonuçların iyileştirilmesine giderek daha fazla vurgu yapılmaktadır.
İleri Okuma
- Lichtenstein, L. (1953). Histiocytosis X: Integration of Eosinophilic Granuloma of Bone, Letterer-Siwe Disease, and Schüller-Christian Disease as Related Manifestations of a Single Nosologic Entity. Archives of Pathology, 56, 84-102.
- Aricò, M., Egeler, R. M. (1998). Clinical Aspects of Langerhans Cell Histiocytosis. Hematology/Oncology Clinics of North America, 12(2), 247-258.
- Weitzman, S., Egeler, R. M. (2005). Histiocytic Disorders of Children and Adults: Introduction to the Problem, Overview, and Classification. Hematology/Oncology Clinics of North America, 19(1), 1-7.
- Allen, C. E., Li, L., Peters, T. L., et al. (2010). MAPK Pathway Activation in Langerhans Cell Histiocytosis. Blood, 116(22), 2256-2262.
- Badalian-Very, G., Vergilio, J. A., Degar, B. A., et al. (2010). Recurrent BRAF Mutations in Langerhans Cell Histiocytosis. Blood, 116(11), 1919-1923.
- Aricò, M., Girschikofsky, M., Généreau, T., et al. (2013). Langerhans Cell Histiocytosis in Adults: Report from the International Registry of the Histiocyte Society. European Journal of Cancer, 49(11), 2758-2767.
- Berres, M. L., Lim, K. P., Peters, T., et al. (2014). BRAF-V600E Expression in Langerhans Cell Histiocytosis Correlates with High-Risk Neurodegenerative Disease. Nature Medicine, 20(7), 845-853.
- Rodriguez-Galindo, C., Allen, C. E. (2014). Langerhans Cell Histiocytosis. Blood, 126(26), 465-472.
- Allen, C. E., Ladisch, S., McClain, K. L. (2015). How I Treat Langerhans Cell Histiocytosis. Blood, 126(1), 26-35.
- Diamond, E. L., Subbiah, V., Lockhart, A. C., et al. (2019). Vemurafenib for BRAF V600–Mutant Erdheim-Chester Disease and Langerhans Cell Histiocytosis: Analysis of Data From the Histology-Independent, Phase 2, Open-label VE-BASKET Study. JAMA Oncology, 5(3), 519-522.