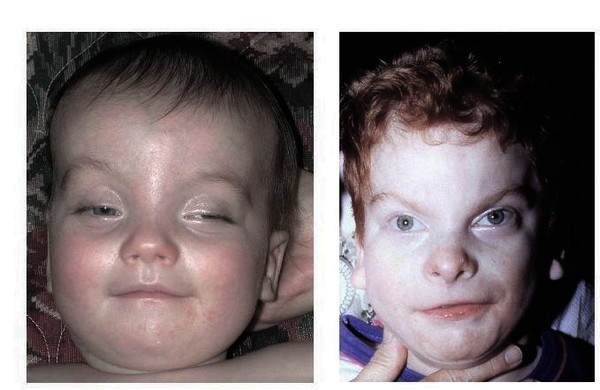
Miller-Dieker Sendromu (MDS) da dahil olmak üzere Lissensefali, normal girus eksikliği (agiri) veya daha az sayıda, daha geniş girus (pakigiri) nedeniyle düz bir beyin yüzeyi ile karakterize nadir bir doğuştan beyin bozukluğudur. Erken fetal gelişim sırasında (gebeliğin 2. ve 4. ayları arasında) nöronal göçün başarısız olmasından kaynaklanır. Bu bozukluk önemli beyin malformasyonlarına ve ciddi nörolojik bozukluklara yol açar.
Neden
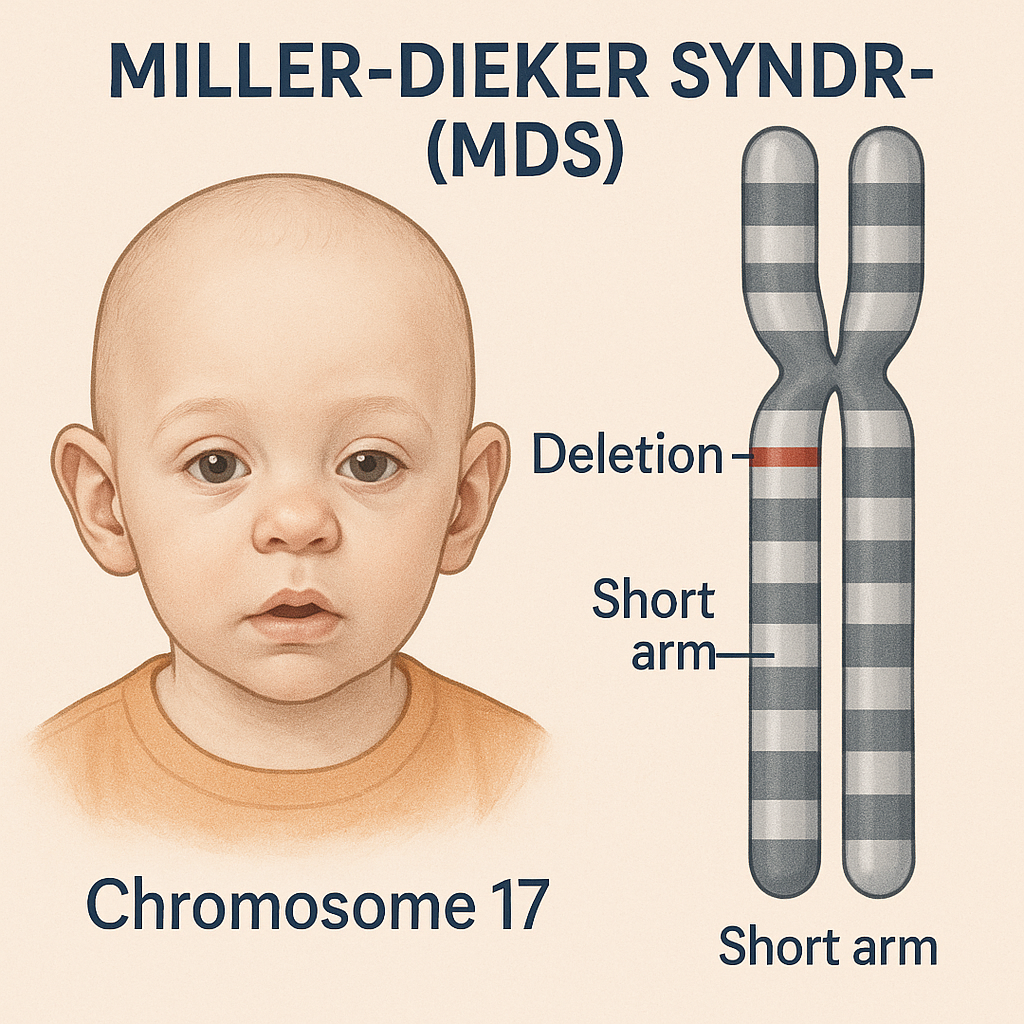
MDS çoğunlukla kromozom 17 (17p13.3) üzerindeki *PAFAH1B1* (eski adıyla LIS1) geni gibi uygun nöronal göç için kritik genlerin silinmesini içeren bir mikrodelesyondan kaynaklanır. Diğer vakalar kortikal gelişimden sorumlu genlerdeki mutasyonlardan kaynaklanmaktadır. Viral enfeksiyonlar, toksik maruziyetler ve genetik mutasyonlar gibi faktörler de MDS’nin başlamasına katkıda bulunur.
Klinik Özellikler:
- Nörolojik ve Beyin Malformasyonları**:
- Lissensefali (pürüzsüz beyin yüzeyi)
- Agiri (girus yokluğu) ve pakigiri (anormal derecede geniş girus)
- Kalınlaşmış serebral korteks
- Diğer Belirtiler**:
- Ciddi zihinsel ve bilişsel bozukluk
- Psikomotor gerilik
- Epilepsi ve spastik nöbetler
- Ciddi konuşma bozuklukları
- Görsel ve işitsel bozukluklar
- Dispne (nefes darlığı) ve tekrarlayan pnömoni dahil solunum sorunları
- Beslenme ve yutma güçlükleri
- Kas sertliği ve spastisite
- Kalp, böbrek ve diğer iç organ malformasyonları da yaygındır.
Teşhis
- Klinik Değerlendirme**: Tanı tipik olarak mikrosefali, hipotoni (kas tonusunun azalması) ve yüz dismorfizmi (ayırt edici yüz özellikleri) gibi görünür fiziksel semptomlara dayanarak konur.
- Görüntüleme**: Beyin malformasyonlarını ve yapısal anomalileri, özellikle de girus yokluğunu tanımlamak için MR ve BT taramaları kullanılır.
- Genetik Test: Kromozomal mikroarray ve hedefe yönelik genetik testler (örneğin LIS1 ve YWHAE için) 17p13.3 kromozomundaki delesyonları veya mutasyonları doğrular.
Tedavi ve Prognoz
- Tedavi Yok**: MDS yapısal bir beyin malformasyonu olduğundan, altta yatan durumun tedavisi yoktur. Tedavi, semptomları yönetmeye ve yaşam kalitesini artırmaya odaklanır.
- Semptom Yönetimi**:
- Nöbetleri kontrol etmek için antiepileptik ilaçlar
- Tekrarlayan pnömoni için solunum desteği ve tedaviler
- Motor fonksiyon bozukluğu ve spastisiteyi ele almak için fiziksel ve mesleki terapiler
- Ciddi yutma güçlüğü olanlar için beslenme tüpleri de dahil olmak üzere beslenme desteği
- Prognoz**: Prognoz genellikle kötüdür. Etkilenen çocukların çoğu, şiddetli solunum yolu enfeksiyonları ve nörolojik işlev bozukluğu gibi komplikasyonlar nedeniyle genellikle ilk birkaç ay ila yıl içinde erken ölüm yaşar.
Yaygın Olmayan Özel Formları
Borth Sendromu:
- Bu lissensefali varyantı Miller-Dieker sendromu ile aynı özellikleri paylaşır ancak benzersiz bir genetik mutasyon veya yapısal beyin modeli sunar. Borth Sendromuna özgü ayrıntılar azdır ancak semptomların şiddetinin değiştiğini göstermektedir.
Norman-Roberts Sendromu:
- MDS’ye benzer şekilde, Norman-Roberts sendromu nöronal göçü etkileyen mutasyonlardan kaynaklanır, ancak farklı yüz anormallikleri (geniş burun köprüsü ve anteverted burun delikleri gibi), şiddetli lissensefali ve zihinsel engellilik ile kendini gösterir. Bu sendrom da sıklıkla 17. kromozomda benzer bir mikrodelesyon içerir.
Fukuyama Sendromu:
- Bu sendrom öncelikle konjenital musküler distrofi ile karakterizedir ve lissensefali ile bağlantılıdır. MDS’den farklı olarak, ciddi gelişimsel gecikmeler ve benzer bilişsel eksikliklerle birlikte daha belirgin kas dejenerasyonu da içerir.
Keşif
Miller-Dieker Sendromu (MDS)** ve altında yatan neden olan lissensefalinin tarihi ve anlaşılması, klinik genetik, nörobilim ve gelişimsel biyolojideki önemli dönüm noktalarıyla gelişmiştir.

İçindekiler
Erken Gözlemler ve İlk Tanımlamalar (1960’lar-1970’ler)
1963 – James Q. Miller’ın İlk Tanımı:
- Dr. James Q. Miller ilk olarak otopsi çalışmalarında görülen pürüzsüz beyin yüzeyinin (lissensefali) yanı sıra ciddi zihinsel engeller, epilepsi ve ayırt edici yüz özellikleriyle karakterize nadir bir durumu olan bir grup hastayı tanımladı.
- Miller, beynin yapısal anormalliklerine odaklanmış ve bu anormallikleri ciddi nörolojik eksikliklerle ilişkilendirmiştir.
1969 – H. Dieker’in Katkısı:
- Dr. H. Dieker, iç organların anormal gelişimi de dahil olmak üzere beynin ötesinde çoklu malformasyonlarla ilişkisi nedeniyle bozukluğun bir sendrom olarak sınıflandırılması gerektiğini vurgulayarak Miller’ın gözlemlerini genişletti.
- Dieker, durumu benzersiz bir sendrom olarak tanımlamış ve lissensefali (düz beyin) ile yüz dismorfizmleri ve diğer gelişimsel malformasyonların kombinasyonunu vurgulamış, böylece “Miller-Dieker Sendromu” (MDS) terimi ortaya çıkmıştır.
1970’ler – Otozomal Resesif Kalıtım Hipotezi:
- İlk genetikçiler, etkilenen ailelerin klinik gözlemlerine dayanarak Miller-Dieker Sendromunun otosomal resesif kalıtım modeli izlediğini varsaymışlardır. Ancak, henüz spesifik bir gen veya kromozomal bölge tanımlanmamıştı.
Genetik Anlayıştaki Gelişmeler (1980’ler-1990’lar)
1980’ler – Lissensefali Üzerine Artan Odaklanma:
- Gelişmiş nörogörüntüleme teknikleri (MRI ve CT gibi) daha yaygın olarak kullanılabilir hale geldikçe, klinisyenler yaşayan hastalarda karakteristik beyin malformasyonlarını görselleştirebildiler. Bu, özellikle Miller-Dieker Sendromunda görülen yüz ve organ özelliklerine sahip hastalarda lissensefalinin daha iyi sınıflandırılmasına ve teşhis edilmesine yol açtı.
- Araştırmacılar lissensefaliyi beyin gelişimi sırasında nöronal göçün başarısız olmasıyla ilişkilendirerek daha ileri genetik analizler için zemin hazırladılar.
1991 – Kromozom 17p13.3 Delesyonunun Tanımlanması:
- Çok önemli bir keşifte, araştırmacılar MDS’li birkaç hastada 17p13.3** kromozomu üzerinde bir mikrodelesyon tespit ettiler. Bu, hastalıktan sorumlu genetik lokusu kesin olarak belirlediği için çığır açan bir andı.
- Kromozomal çalışmalar, bu mikrodelesyonun şu anda LIS1 (veya PAFAH1B1) olarak bilinen ve beyin gelişimi sırasında nöronal göçün düzenlenmesinde çok önemli bir rol oynayan bir geni kapsadığını ortaya koydu.
1993 – LIS1 ve Lissensefali Arasındaki Bağlantı:
- Daha ileri çalışmalar, LIS1 genindeki mutasyonlar veya delesyonlar ile lissensefalide, özellikle de MDS hastalarında görülen kortikal malformasyonlar arasında doğrudan bir bağlantı kurmuştur.
- Araştırmacılar, LIS1 proteininin uygun nöronal göç için, özellikle de serebral korteksi oluşturmak için göç ederken nöronlardaki hücre iskeleti dinamiklerini düzenlemede gerekli olduğunu belirlediler.
Modern Gelişmeler (2000’ler-Günümüz)
2000’ler – Nöronal Göç Bozukluklarını Anlamak:
- Devam eden araştırmalar, MDS ve izole lissensefali sekansı (ILS) gibi diğer ilgili durumlar da dahil olmak üzere nöronal migrasyon bozukluklarının (NMD’ler) anlaşılmasını genişletti. LIS1 geni** nöronal hareketin kilit düzenleyicilerinden biri olarak ortaya çıktı ve eksikliği beyin malformasyonlarının ciddiyetiyle ilişkilendirildi.
- Bu dönemde, araştırmacılar kromozom 17p13.3 üzerinde YWHAE gibi silindiğinde veya mutasyona uğradığında Miller-Dieker Sendromunun şiddetini artırabilen veya değiştirebilen ve farklı fenotipik ifadelere yol açan diğer genleri tanımladılar.
2010’lar – Teşhis ve Tedavide Gelişmeler:
- Yüksek çözünürlüklü genetik testlerin** (örn. kromozomal mikroarray ve yeni nesil dizileme) kullanılabilirliği ile klinisyenler LIS1 ve diğer ilgili genlerdeki daha küçük delesyonları veya nokta mutasyonlarını daha doğru bir şekilde tanımlayabilmiş ve bu da MDS tanısının iyileştirilmesine yol açmıştır.
- Hedefe yönelik tedaviler geliştirme çabaları epilepsi, beslenme güçlükleri ve solunum sorunları gibi semptomları yönetmeye odaklanmıştır. MDS için bir tedavi bulunamamış olsa da, erken müdahale ve destekleyici bakım bazı hastaların yaşam kalitesini iyileştirmiştir.
2020’ler – Genetik ve Terapötik Araştırmaların Genişletilmesi:
- Devam eden araştırmalar, nöronal hücrelerdeki LIS1 fonksiyon eksikliğini telafi etmeyi amaçlayan gen terapilerini ve diğer moleküler stratejileri keşfetmeye devam etmektedir. Bilim insanları ayrıca Miller-Dieker Sendromu ve ilgili bozuklukların şiddetini etkilemek için genetik yatkınlıkla etkileşime girebilecek çevresel faktörlerin (örn. enfeksiyonlar, toksinler) rolünü araştırmaktadır.
Genişletilmiş Genetik Bilgiler
Araştırmalar, MDS’ye yol açan kromozomal delesyonların veya mutasyonların genellikle LIS1 ve uygun nöronal konumlandırma için gerekli diğer düzenleyici genler dahil olmak üzere birden fazla geni kapsadığını göstermiştir. Ayrıca, daha kapsamlı delesyonlara sahip bireyler, özellikle beyin dışı sistemleri etkileyen daha geniş bir malformasyon yelpazesi sergileyebilir.
Bazı araştırmacılar, belirli çevresel faktörler (örneğin, hamilelik sırasında viral enfeksiyonlar) ve genetik yatkınlık arasındaki etkileşimlerin sendromun fenotipik şiddetini daha da kötüleştirebileceğine inanmaktadır. Dolayısıyla MDS, nörogelişimsel bozukluklar üzerinde hem genetik hem de çevresel etkilerin önemini örneklemektedir.
İleri Okuma
- Miller, J.Q. (1963). A case of lissencephaly with severe intellectual disability. Neurology, 13, 841-845.
- Dieker, H.E. (1969). Lissencephaly syndrome: A clinical and neuropathological study of a congenital disorder. Journal of Pediatrics, 75, 245-258.
- Dobyns, W.B., et al. (1995). Clinical findings in patients with lissencephaly and subcortical band heterotopia. Journal of Medical Genetics, 32(10), 721-725.
- Dobyns, W. B., & Truwit, C. L. (1995). Lissencephaly and other malformations of cortical development: 1995 update. Neuropediatrics, 26(3), 132–147.
- Pilz, D. T., & Quarrell, O. W. J. (1996). LIS1 and the genetics of lissencephaly. Current Opinion in Neurology, 9(2), 117–121.
- Chong, S.S., et al. (1997). Deletion of LIS1 and 17p13.3 in patients with Miller-Dieker syndrome. Human Molecular Genetics, 6(1), 147-155.
- Chong, S. S., Pack, S. D., Roschke, A. V., Tanigami, A., Carrozzo, R., Smith, A. C., & Ledbetter, D. H. (1997). A revision of the lissencephaly and Miller-Dieker syndrome critical regions in chromosome 17p13.3. Human Molecular Genetics, 6(1), 147–155.
- Pilz, D.T., et al. (1998). LIS1 and lissencephaly: Molecular genetics and pathophysiology. Human Molecular Genetics, 7(13), 2029-2036.
- Gleeson, J. G., Allen, K. M., Fox, J. W., Lamperti, E. D., Berkovic, S., Scheffer, I., & Dobyns, W. B. (1998). Lissencephaly and subcortical band heterotopia: Molecular basis and diagnosis. Brain, 121(11), 2059–2070.
- Reiner, O., & Sapir, T. (1999). LIS1: Molecular pathways and signaling mechanisms involved in neuronal migration and lissencephaly. Journal of Neurobiology, 39(4), 533-544.
- Ross, M.E., & Walsh, C.A. (2001). Human brain malformations and their links to developmental neurogenetics. Annual Review of Neuroscience, 24, 1041-1074.
- Mochida, G.H., & Walsh, C.A. (2004). Genetic aspects of lissencephaly and its diagnosis. Developmental Medicine & Child Neurology, 46(8), 587-592.
- Wynshaw-Boris, A. (2007). Lissencephaly and other malformations of cortical development: Molecular basis and mechanisms. Human Molecular Genetics, 16(2), 184-191.
- Guerrini, R., & Parrini, E. (2010). Neuronal migration disorders. Neurobiology of Disease, 38(2), 154–166.
- Guerrini, R., & Dobyns, W.B. (2014). Malformations of cortical development: Clinical features and genetic causes. Lancet Neurology, 13(7), 710-726.