
Talasemi, Yunanca “thalassa ‘ (’deniz” anlamına gelir) ve “anemi ” (kan eksikliği veya yetersiz kırmızı kan hücresi anlamına gelir) kelimelerinden kaynaklanan genetik bir hemoglobin üretim bozukluğudur. Bu isim, hastalığın Akdeniz toplumlarında, özellikle de denize yakın bölgelerde yüksek prevalansını yansıtmaktadır. Talasemi, hemoglobin molekülünü oluşturan protein zincirlerinden birinde (alfa veya beta) eksiklikle karakterize olup, asemptomatikten şiddetli, hayatı tehdit eden anemiye kadar çeşitli klinik belirtilere yol açar.
Talaseminin Etimolojisi
Talasemi** terimi 20. yüzyılın başlarında Yunanca’dan türetilmiştir:
- Thalassa**: “Deniz” anlamına gelir ve bu durumun ilk kez tanımlandığı Akdeniz bölgesine atıfta bulunur.
- Anemi**: Durumun ayırt edici özelliği olan yeterli kırmızı kan hücresi veya hemoglobin eksikliğini ifade eder.
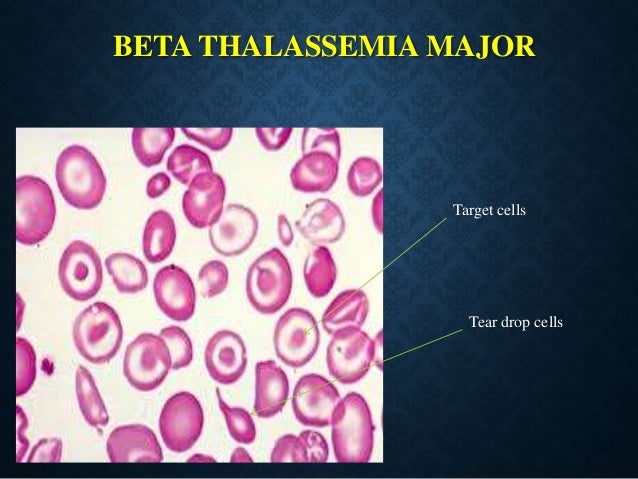
Talasemi Epidemiyolojisi
Talasemi özellikle tropikal ve subtropikal bölgelerde, özellikle de tarihsel olarak sıtmaya maruz kalmış popülasyonlarda yaygındır. Bu durum Akdeniz, Afrika, Orta Doğu, Hint Yarımadası ve Güneydoğu Asya’da oldukça yaygındır. Bu bölgelerdeki yaygınlığının nedeni, talasemi taşıyıcılarının sıtmaya karşı bir miktar korumaya sahip olduğu ve bu popülasyonlarda özelliğin daha yüksek bir sıklığa yol açtığı sıtma hipotezi ile ilgilidir.
- Küresel yaygınlık**: Küresel nüfusun yaklaşık *%1,5’i* (90 milyondan** fazla kişi) β-talasemi özelliği taşımaktadır.
- Ağır vakaların doğum oranı**: Her yıl dünya çapında *60.000 ila 80.000 ağır talasemi vakasının* doğduğu tahmin edilmektedir.
Talaseminin Klinik Belirtileri
Talasemi, şiddetine bağlı olarak farklı şekillerde ortaya çıkar. Hastalık klinik tabloya göre üç ana forma ayrılır:
Talasemi Minör (Kalıtsal):
- Genellikle asemptomatik veya hafif anemi ile seyreder.
- Bireyler taşıyıcıdır ancak tipik olarak tedavi gerektirmezler.
Talasemi Major (Cooley Anemisi):
- Bebeklik döneminde başlayan şiddetli anemi.
- Belirtiler arasında büyüme ve gelişmede gecikme, iskelet anormallikleri (özellikle yüz ve kafatasında), dalak büyümesi ve sarılık yer alır.
- Hastalar, transfüzyonlardan kaynaklanan aşırı demir yükünü yönetmek için ömür boyu kan transfüzyonu ve şelasyon tedavisi gerektirir.
Talasemi İntermedia:
- Semptomlar talasemi minör ve majör arasında orta şiddette seyreder.
- Bu bireylerde hafif ila şiddetli anemi, büyümede yavaşlama ve kemik anormallikleri olabilir.
- Tedavi semptomların ciddiyetine bağlıdır ve ara sıra kan nakli ve demir şelasyon tedavisi içerebilir.
Talasemi Türleri
Talasemi, etkilenen globin zincirine (alfa veya beta) ve ilgili gen mutasyonlarının sayısına göre sınıflandırılır.
İçindekiler
Alfa Talasemi
Alfa talasemi, hemoglobinin alfa globin zincirlerini kodlayan HBA1 ve HBA2 genlerindeki mutasyonlardan kaynaklanır. Kusurlu genlerin sayısına göre sınıflandırılan dört tip alfa talasemi vardır:
- Sessiz Taşıyıcı: Bir kusurlu alfa geni; semptom yok.
- Alfa Talasemi Minör (Özellik): İki kusurlu alfa geni; hafif anemi.
- Hemoglobin H Hastalığı: Üç kusurlu alfa geni; orta ila şiddetli anemi, dalak büyümesi ve kemik deformiteleri.
- Alfa Talasemi Major (Hidrops Fetalis): Dört kusurlu alfa geni; bu durum tedavi edilmezse rahimde veya doğumdan kısa bir süre sonra ölümcüldür.
Beta Talasemi
Beta talasemi, hemoglobinin beta globin zincirini kodlayan HBB genindeki mutasyonlardan kaynaklanır. İki ana formu vardır:
- Beta Talasemi Major (Cooley Anemisi): Beta globin üretiminin tamamen olmaması şiddetli anemiye yol açar. Hastalar ömür boyu kan nakline ve şelasyon tedavisine ihtiyaç duyar.
- Beta Talasemi Minör (Özellik): Beta globin üretiminin azalması, genellikle tedavi gerektirmeyen hafif anemiye yol açar.
Alfa ve beta talasemiye ek olarak, hemoglobinin delta ve gama zincirlerini etkileyen delta ve gamma talasemi gibi nadir formlar da vardır. Bu formlar da spesifik genlerdeki mutasyonları içerir ve genetik testler ve laboratuvar yöntemleri kullanılarak teşhis edilir.
Global Tanı Kriterleri
Talasemi tanısı tipik olarak aşağıdaki testleri içerir:
- Tam Kan Sayımı (CBC)**: Anemi ve kırmızı kan hücresi anormalliklerini kontrol etmek için.
- Hemoglobin Elektroforezi**: Beta talasemide yüksek fetal hemoglobin (HbF) ve hemoglobin A2 (HbA2) seviyeleri gibi mevcut hemoglobin türlerini ve miktarlarını belirlemek için.
- DNA Analizi**: Kesin tanı için, alfa veya beta globin genlerindeki mutasyonları veya delesyonları tanımlamak üzere genetik testler kullanılır.
Tedavi Kılavuzları
Talaseminin tedavisi ciddiyetine bağlıdır:
Talasemi Minör:
- Genellikle tedavi gerektirmez. Hastalar tipik olarak asemptomatiktir ve sadece hafif anemileri olabilir.
Talasemi Major:
- Düzenli kan transfüzyonları**: Hemoglobin seviyelerini korumak ve normal büyüme ve gelişmeyi sağlamak için gereklidir.
- Demir şelasyon tedavisi**: Düzenli kan nakillerinden kaynaklanan aşırı demir yüklenmesini önlemek için verilir. Fazla demirin vücuttan atılması için *deferoksamin* veya deferasiroks gibi ilaçlar kullanılır.
- Folik asit takviyeleri**: Kırmızı kan hücresi üretimini desteklemek için önerilebilir.
- Kemik iliği veya kök hücre nakli**: Bu, uygun bir donör mevcutsa şiddetli talasemi için tek potansiyel tedavidir.
Talasemi İntermedia:
- Tedavi semptomların şiddetine bağlı olarak değişir ancak ara sıra kan transfüzyonu ve demir şelasyon tedavisini içerebilir.
Son Gelişmeler ve Gelecekteki Yönelimler
- Gen Terapisi: Talasemi için gen terapisi araştırmaları, altta yatan genetik kusuru düzeltmek ve potansiyel olarak hastalığı iyileştirmek amacıyla devam etmektedir.
- Geliştirilmiş Demir Şelasyon Tedavileri**: Şelasyon ilaçlarındaki gelişmeler, aşırı demir yükünün yükünü azaltarak hastaların yaşam kalitesini artırmıştır.
- Doğum Öncesi Test ve Genetik Danışmanlık**: Talasemi prevalansının yüksek olduğu bölgelerde, doğum öncesi genetik test ve danışmanlık, hastalığın erken teşhisi ve yönetimi için önemlidir.
Keşif
1904 – Talaseminin İlk Tanımı
- Yunan doktor Thomas Bizzozero şiddetli anemi ve kemik deformiteleri olan çocuk vakalarını belgeleyerek talaseminin bilinen ilk tanımını yaptı.
1925 – Talaseminin İsimlendirilmesi
- Dr. George Whipple ve Dr. Thomas Cooley Akdeniz kökenli çocuklarda ciddi bir anemi türü tespit etmiştir. Daha sonra beta-talasemi major olarak bilinen Cooley anemisine Dr. Cooley’in adı verilmiştir.
1932 – “Talasemi” Terimi Ortaya Çıktı
- Talasemi** terimi Dr. George Whipple tarafından ortaya atılmış olup, Yunanca “thalassa” (deniz) ve “anemi” kelimelerinden türetilmiştir ve hastalığın Akdeniz toplumlarındaki yüksek prevalansını yansıtmaktadır.
1949 – Talasemi için Kan Transfüzyonu
- Talasemi majör tedavisi olarak düzenli kan transfüzyonlarının ilk kullanımı başlatıldı ve şiddetli anemi yönetiminde bir köşe taşı haline geldi.
1960’lar – Demir Şelasyon Tedavisinin Geliştirilmesi
- Sık kan transfüzyonlarından kaynaklanan aşırı demir yükünü tedavi etmek için Demir şelasyon tedavisi kullanılmaya başlandı. Bu, talasemi hastalarının uzun vadeli yönetiminde önemli bir gelişmeye işaret ediyordu.
1970’ler – Talasemide Fetal Hemoglobinin Keşfi
- Araştırmacılar fetal hemoglobin (HbF) üretiminin beta-talasemide semptomların şiddetini azaltabileceğini keşfederek HbF seviyelerini artırmaya yönelik terapötik stratejilerde ilerlemelere yol açtı.
1980’ler – Prenatal Tanı ve Genetik Danışmanlık
- Genetik testlerin kullanıldığı Prenatal tanı geliştirilerek fetüslerde talaseminin erken teşhisi sağlandı ve aile planlaması ile bilinçli karar verme süreçlerine olanak tanındı.
2000’ler – Gen Terapisi Araştırmaları
- Altta yatan genetik kusuru düzelterek talasemiyi iyileştirmeyi amaçlayan gen terapisi araştırmalarındaki ilerlemeler, gelecekteki iyileştirici tedaviler için umut vaat ediyor.
2010’lar – Geliştirilmiş Şelasyon İlaçları
- Önceki tedavilere kıyasla daha kolay oral uygulama ve daha iyi uyum sunan deferasiroks gibi yeni demir şelatlama ajanları geliştirildi.
2019 – Beta-Talasemi için Gen Tedavisinin Onaylanması
- Avrupa İlaç Ajansı, beta-talasemi hastaları için bir gen terapisi olan Zynteglo‘yu onaylayarak hastalığın iyileştirici tedavilerinde büyük bir atılım gerçekleştirdi.
Yürütülmekte Olan Araştırma – Gen Düzenleme
- CRISPR-Cas9** ve diğer gen düzenleme teknolojileri, talasemi için potansiyel iyileştirici tedaviler olarak araştırılmakta ve klinik deneyler umut verici erken sonuçlar göstermektedir.
Bu kilometre taşları, geçtiğimiz yüzyılda talaseminin teşhisi, tedavisi ve potansiyel tedavisinde kaydedilen önemli ilerlemeleri göstermektedir.
İleri Okuma
- Olivieri, N. F., & Brittenham, G. M. (1997). Iron-chelating therapy and the treatment of thalassemia. Blood, 89(3), 739-761.
- Cao, A., Galanello, R., & Rosatelli, M. C. (1997). Prenatal diagnosis and screening of the hemoglobinopathies. Baillière’s Clinical Haematology, 10(2), 203-222.
- Rund, D., & Rachmilewitz, E. (2005). Beta-thalassemia. New England Journal of Medicine, 353(11), 1135-1146.
- Vichinsky, E. P. (2005). Changing patterns of thalassemia worldwide. Annals of the New York Academy of Sciences, 1054(1), 18-24.
- Modell, B., & Darlison, M. (2008). Global epidemiology of haemoglobin disorders and derived service indicators. Bulletin of the World Health Organization, 86, 480-487.
- Cao, A., & Galanello, R. (2010). Beta-thalassemia. Genetics in Medicine, 12(2), 61-76.
- Galanello, R., & Origa, R. (2010). Beta-thalassemia. Orphanet journal of rare diseases, 5(1), 1-12.
- Galanello, R., & Origa, R. (2010). Alpha-thalassemia. Orphanet Journal of Rare Diseases, 5(1), 1-15.
- Weatherall, D. J. (2010). The inherited diseases of hemoglobin are an emerging global health burden. Blood, 115(22), 4331-4336.
- Borgna-Pignatti, C., & Gamberini, M. R. (2011). Complications of thalassemia major and their treatment. Expert Review of Hematology, 4(3), 353-366.
- Cappellini, M. D., Cohen, A., Porter, J., Taher, A., & Viprakasit, V. (Eds.). (2014). Guidelines for the management of transfusion dependent thalassemia (TDT) (3rd ed.). Thalassemia International Federation.
- Angelucci, E., Matthes-Martin, S., Baronciani, D., Bernaudin, F., Zecca, M., Maury, S., & Locatelli, F. (2014). Hematopoietic stem cell transplantation in thalassemia major. Haematologica, 99(5), 833-840.
- Origa, R. (2017). Beta-Thalassemia. Genereviews.
- Lal, A., & Goldrich, M. L. (2020). Alpha Thalassemia. In StatPearls [Internet]. StatPearls Publishing.
- Taher, A. T., Musallam, K. M., & Cappellini, M. D. (2021). Thalassemia. The Lancet, 397(10291), 369-384.