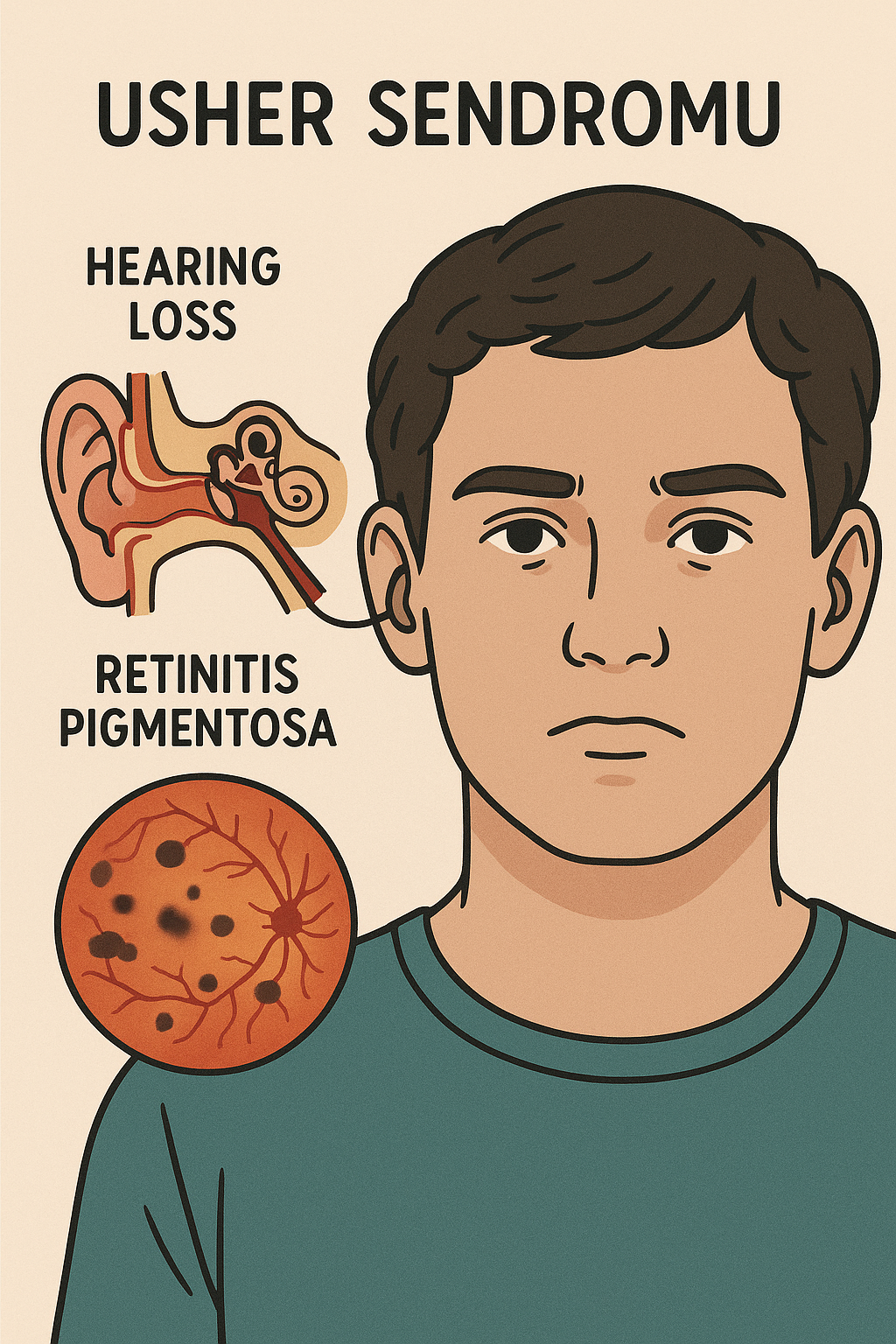
Usher sendromu iyi karakterize edilmiş bir genetik bozukluk ve siliopati‘nin klasik bir örneğidir. İnsanlarda birleşik sağırlık ve körlüğün en yaygın nedenidir ve bazı durumlarda denge üzerinde ek etkilere sahiptir.
İçindekiler
Usher Sendromunun Temel Özellikleri
Birincil Semptomlar:
- Sensorinöral işitme kaybı: Koklear saç hücrelerindeki kusurlar nedeniyle doğuştan (konjenital) mevcuttur.
- Retinitis pigmentosa (RP): Görme kaybına yol açan retina fotoreseptörlerinin ilerleyici dejenerasyonu (önce gece körlüğü, sonra tünel görüşü ve potansiyel olarak tam körlük).
- Vestibüler disfonksiyon: Bazı alt tiplerde denge sorunları (örn. Usher tip 1).
Alt tipler:
- Tip 1 (USH1):
- Derin doğuştan sağırlık, ciddi denge sorunları ve erken başlangıçlı RP (görme kaybı çocuklukta başlar).
- MYO7A, USH1C veya CDH23 gibi genlerdeki mutasyonlardan kaynaklanır.
- Tip 2 (USH2):
- Orta ila şiddetli doğuştan işitme kaybı (denge sorunu yok), RP ergenlik/yetişkinlikte başlar.
- En yaygın alt tip (vakaların yaklaşık %50’si), USH2A (usherin) veya ADGRV1 mutasyonlarıyla bağlantılıdır.
- Tip 3 (USH3):
- İlerleyen işitme kaybı ve değişken RP başlangıcı; denge sorunları gelişebilir.
- CLRN1 mutasyonlarıyla ilişkilidir.
Siliopatilerle Bağlantı
Usher sendromu, patolojisi işlevsiz birincil silyalar veya siliopatiyle ilişkili yapılar içerdiğinden siliopati olarak sınıflandırılır:
- İç kulak kıl hücreleri: Stereosilyalar (aktin tabanlı çıkıntılar, gerçek silyalar değil) uygun yapı ve mekanotransdüksiyon için Usher proteinlerine ihtiyaç duyar.
- Retinal fotoreseptörler: Bağlantı silyası (değiştirilmiş birincil silya), proteinleri ışığa duyarlı dış segmentlere taşımak için kritik öneme sahiptir. Kusurlar fotoreseptör sağkalımını bozar.
Usher protein kompleksleri (örn. USH1/USH2 proteinleri) silyalarla ilgili süreçlerde moleküler köprüler görevi görür, bunlara şunlar dahildir:
- Stereosilya demetlerinin yapısal kohezyonu.
- Fotoreseptör silyalarının bakımı.
- Sinyal yolları (örn. Hedgehog, Wnt).
Genetik ve Moleküler Temel
- Otozomal resesif kalıtım: Genin her iki kopyası da mutasyona uğramış olmalıdır.
- Anahtar genler:
- MYO7A (USH1B): Stereosilya işlevi için kritik olan aktin bazlı bir motor proteini kodlar.
- USH2A: Fotoreseptör silyum ve kokleadaki bir protein olan usherin’i kodlar.
- CLRN1 (USH3A): Sinaptik işlev ve silya bakımında rol oynar.
Araştırma ve Terapötik Yaklaşımlar
- Gen Terapisi: İşlevsel genleri retinaya/iç kulağa iletmek için viral vektörler kullanan MYO7A (USH1B) ve USH2A‘yı hedefleyen denemeler.
- Antisense Oligonükleotidler (ASO’lar):Mutasyonları atlamak veya protein fonksiyonunu geri yüklemek (örn. USH2A için).
- CRISPR/Cas9: Klinik öncesi modellerde mutasyonları düzenlemek.
- Retinal Protezler ve Koklear İmplantlar: Görme/işitme kaybı için semptomatik yönetim.
- Siliopati: Usher sendromu prototipik bir örnektir.
- BMP40: BMP sinyalizasyonu doğrudan Usher sendromuyla bağlantılı olmasa da, Hedgehog (silya bağımlı) ve Wnt gibi yollar siliyopatilerde bozulur. BMP’ler retinal veya koklear gelişimde dolaylı roller oynayabilir. –
- Tek hücreli organoidler: Usher sendromunu modellemek için kullanılır, saç hücresi veya fotoreseptör dejenerasyonunun incelenmesine ve gen terapilerinin test edilmesine olanak tanır.
- BAR alanları: Bazı Usher proteinleri (örn. harmonin/USH1C), stereosilya zarlarını şekillendirmek için membran kıvrımlı BAR alanı proteinleriyle etkileşime girer.
Güncel Zorluklar
- Heterojenlik: 10’dan fazla gen Usher sendromuna neden olur ve hedefli terapileri zorlaştırır.
- Geç tanı: Görme kaybı yavaş ilerler, genellikle geri döndürülemez hasardan sonra tespit edilir.
- Çift duyusal kayıp: Hem işitme hem de görme eksikliklerinin yönetimi multidisipliner bakım gerektirir.
Keşif
- 1858: Albrecht von Graefe’nin Gözlemleri – Alman göz doktoru Albrecht von Graefe, retinitis pigmentosa (ilerleyen görme kaybı) ve doğuştan sağırlık hastalarını not ederek Usher sendromuna benzeyen bir durumu ilk kez tanımladı. Bu, resmi olarak tanımlanmasının erken bir öncüsü olarak kabul edilir.
- Richard Liebreich (1861): Gräfe’nin öğrencisi Liebreich, Berlin’de yaptığı çalışmada, akraba evliliklerinin yoğun olduğu ailelerde RP ve sağırlık birlikteliklerinin kalıtımsal olduğunu öne sürerek, bu bulguların otozomal resesif geçişi işaret ettiğini belirtmiştir.
Charles Howard Usher’ın Kısa Özgeçmişi
- Doğum ve Eğitim: 2 Mart 1865’te Edinburgh’da doğan Usher, tıp eğitimini Cambridge Üniversitesi ile Londra’daki St. Thomas’s Hospital’da tamamlamış; 1891’de doktorasını almıştır.
- Profesyonel Kariyer: Edward Nettleship’in yanında asistanlık yaptıktan sonra, Aberdeen Royal Infirmary ile Aberdeen Hospital for Sick Children’da göz cerrahı olarak görev yapmış, 1926’da emekli olana dek Skotlandiya’nın kırsal kesimindeki oftalmolojik örüntüleri incelemiştir.
- Diğer Çalışmalar: 1912’de Karl Pearson ile birlikte “A Monograph on Albinism in Man” adlı önemli bir albinoz monografisi yayımlamıştır.

“On the Inheritance of Retinitis Pigmentosa, with Notes of Cases” (1914)
- Çalışmanın Amacı ve Kapsamı: Usher, 1914’te British Journal of Ophthalmology Raportları’nda (“Roy. Lond. Ophthalmol. Hosp. Rep.”, cilt 19, s. 130–236) yayımladığı makalesinde, 69 RP’li bireyi kapsayan bir olgu serisini sistematik olarak incelemiştir. Bu bireyler 40 ayrı aileden seçilmiştir.
- Olguların Seçimi ve İncelenen Bulgular: 69 RP hastasının her birinde ayrıntılı oftalmoskopik muayene, görme alanı testleri ve anamnez yoluyla işitme durumu sorgulanmıştır. Usher, bu bireylerden 19’unun doğuştan işitme kaybı taşıdığını; diğerlerinde ise işitme fonksiyonlarının nispeten korunduğunu tespit etmiştir.
- Kalıtsal Model ve Pedigri Analizi: Aile öyküleri derlenerek pedigriler oluşturulmuş; hem anne hem baba tamamen sağlıklı görünürken, birden fazla çocuğun RP ve/veya sağırlıkla dünyaya gelmesi otozomal resesif geçişi kuvvetle desteklemiştir. Usher, bu kalıtım modelini ilk kez bu kadar kapsamlı örneklemle belgelemiştir.
Yayın Sonrası Kabul ve Eponimleşme Süreci
- 1914–1935 Dönemi: Makalenin yayımlandığı dönemde, konuyla ilgilenen oftalmolog ve genetikçiler bu kapsamlı çalışmayı hızla referans almış; 1935’te Usher, British Ophthalmological Society’nin Bowman Lecture’ında RP üzerine konuşmuş, ancak burada işitme kaybı bağlantısını tekrarlamamıştır.
- “Usher Sendromu” Kavramının Yerleşmesi: Zamanla, RP ile sağırlığın birlikteliğini anımsatan sendromun adı, çalışmanın yayıncısının soyadıyla anılmaya başlamış; 20. yüzyıl ortalarına gelindiğinde tıbbi literatürde “Usher syndrome” ya da “Usher–Hallgren syndrome” olarak standartlaştırılmıştır.
—
- 1950’ler-1960’lar: Klinik Sınıflandırma Başlıyor – Araştırmacılar, Usher sendromunun alt tiplerini şiddet ve başlangıç temelinde ayırt etmeye başladılar. Tip 1 (doğumda şiddetli sağırlık, erken görme kaybı) ve Tip 2 (orta düzeyde işitme kaybı, daha sonra görme kaybı) gayri resmi olarak kaydedildi, ancak resmi sınıflandırma daha sonra geldi.
- 1977: Resmi Alt Tip Sınıflandırması – Usher sendromu Tip 1 ve Tip 2 arasındaki ayrım, araştırmacılar tarafından sağlamlaştırıldı; Tip 1 şiddetli doğuştan sağırlık ve denge sorunlarıyla, Tip 2 ise vestibüler sorunlar olmadan daha hafif, stabil işitme kaybıyla ilişkilendirildi.
- 1980’ler: Genetik Araştırma Ortaya Çıkıyor – Genetikteki gelişmeler, bilim insanlarının Usher sendromunun kalıtsal mutasyonlardan kaynaklandığı hipotezini ortaya atmalarına olanak sağladı. Çalışmalar, otozomal resesif kalıtım modelini haritalamaya başladı.
- 1994: İlk Gen Tanımlandı (USH1B) – Usher sendromu Tip 1B ile ilişkili olan MYO7A geni, araştırmacılar tarafından keşfedildi ve genetik temelinin anlaşılmasında büyük bir atılım oldu. Bu gen, işitme ve görme için kritik olan miyozin proteinlerini etkiler.
- 1995-2000’ler: Ek Genler Haritalandı – USH2A (Tip 2A, 1998) ve PCDH15 (Tip 1F, 2001) dahil olmak üzere Usher sendromuyla bağlantılı birden fazla gen tanımlandı. Bu, sonunda en az 11 genin duruma bağlanmasıyla genetik heterojenliğinin anlaşılmasını genişletti.
- 2010: Tip 3 Tanınırlık Kazanıyor – Değişken başlangıçlı ilerleyici işitme ve görme kaybıyla karakterize olan Usher sendromu Tip 3, özellikle CLRN1 geninin (USH3A) tanımlanmasıyla daha yaygın bir şekilde incelendi ve ayırt edildi.
- 2010’lar-Günümüz: Terapötik Gelişmeler – Araştırma, gen terapisi denemeleri (örneğin, MYO7A ve USH2A’yı hedef alan) ve retina implantları dahil olmak üzere potansiyel tedavilere doğru kaydı. Klinik denemeler, görme kaybını yavaşlatma konusunda umut vadetmeye başladı, ancak 2025 itibarıyla herhangi bir tedavi mevcut değil.
İleri Okuma
- Von Graefe, A. (1858). Beitrag zur Pathologie und Therapie des Glaukoms. Archiv für Ophthalmologie, 4, 250–273.
- Liebreich, R. (1861). Zur Diagnose der Amaurose bei angeborener Taubstummheit. Archiv für Ophthalmologie, 7, 266–289.
- Usher, C. H. (1914). On the inheritance of retinitis pigmentosa, with notes of cases. Royal London Ophthalmic Hospital Reports, 19, 130–236.
- Davenport, C. B., & Weeks, D. F. (1915). A statistical study of the occurrence of retinitis pigmentosa. Proceedings of the National Academy of Sciences, 1(9), 535–537.
- François, J. (1961). Hereditary tapetoretinal degenerations. The American Journal of Ophthalmology, 51(6), 1029–1065.
- Smith, R. J. H., & Kelley, P. M. (1991). Usher syndrome: Clinical features, molecular genetics, and advances in therapy. Current Opinion in Otolaryngology & Head and Neck Surgery, 1(6), 472–480.
- Hope, C. I., Bundey, S., Proops, D., & Fielder, A. R. (1997). Usher syndrome in the city of Birmingham—Prevalence and clinical classification. The British Journal of Ophthalmology, 81(1), 46–53.
- Petit, C. (2001). Usher syndrome: From genetics to pathogenesis. Annual Review of Genomics and Human Genetics, 2, 271–297.
- Reiners, J., Nagel-Wolfrum, K., Jürgens, K., Märker, T., & Wolfrum, U. (2006). Molecular basis of human Usher syndrome: Deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease. Experimental Eye Research, 83(1), 97–119.
- Mathur, P., & Yang, J. (2015). Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities. Biochimica et Biophysica Acta (BBA) – Molecular Basis of Disease, 1852(3), 406–420.