İçindekiler
Patofizyoloji ve Genetik:
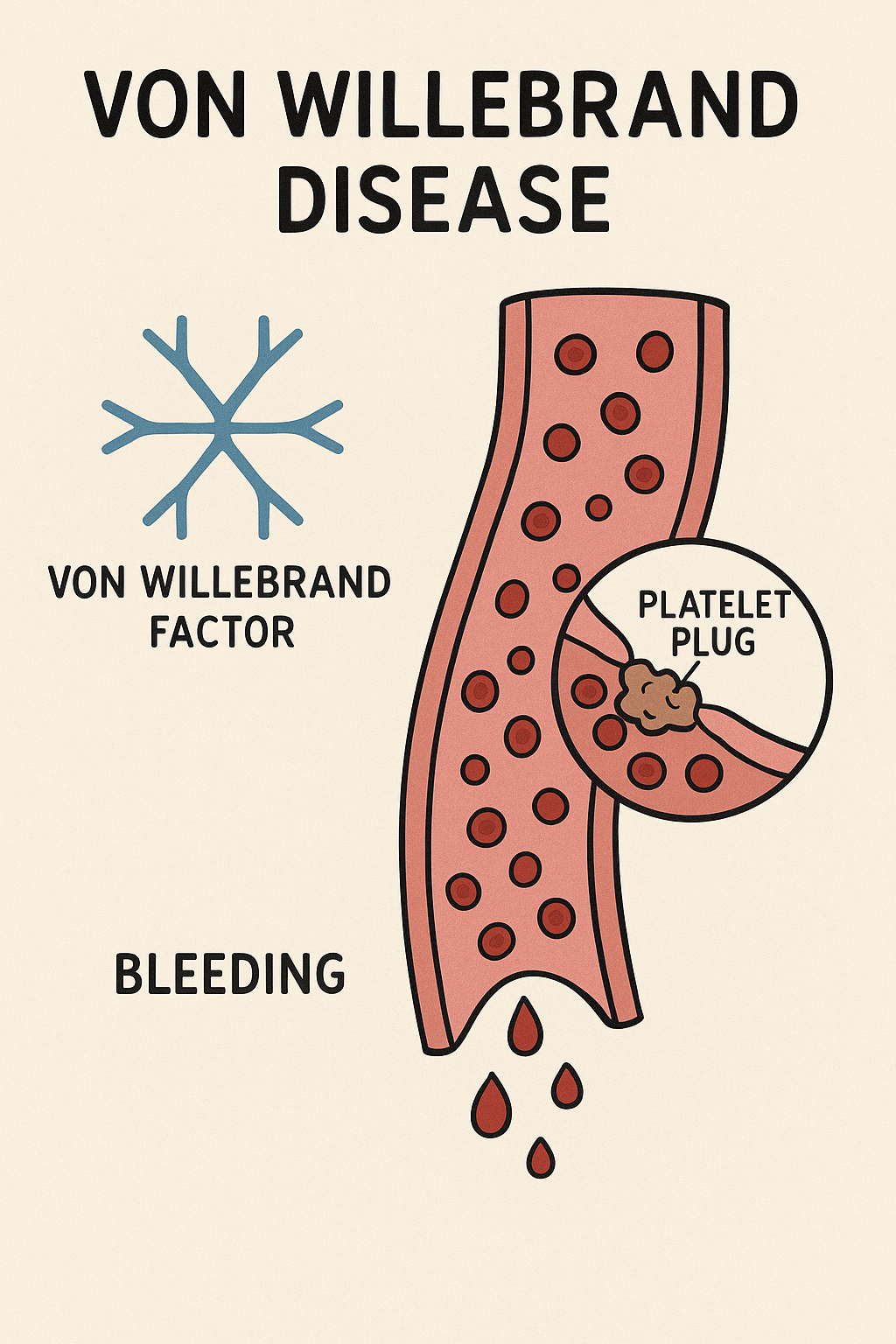
Von Willebrand faktörü (VWF), damar yaralanmasında damar altı subendoteline bağlanarak trombositlerin yapışmasını sağlar ve dolaşımdaki faktör VIII’i stabilize ederek yıkımını önler. VWF, endotelyal hücreler ve megakaryositlerden salgılanan büyük multimerik bir glikoproteindir; kanama durumunda trombositlerin GPIb reseptörü aracılığıyla yapışmasını kolaylaştırır. Faktör VIII ise karaciğerde sentezlenen, A1-A2-B-A3-C1-C2 dizilimli büyük bir koagülasyon ko-faktörüdür; aktif formu (FVIIIa), faktör IXa ile kompleks oluşturarak faktör X’in aktive olmasını sağlar (intrinsik yol). Faktör VIII, plazmada VWF ile sıkıca bağlı dolaşır ve VWF’den ayrıldığında aktif hale gelir.
VWD’nin moleküler temelini VWF genindeki mutasyonlar oluşturur. VWF geni, kromozom 12’nin kısa kolunda (12p13.2) yerleşiktir ve ~178 kb uzunluğunda 52 ekson içerir. Genin genişliğinden ötürü transkripsiyonu ve genetik analizleri zordur; 22q11.2’de VWF’e benzer bir psödogen de bulunmaktadır. VWF genindeki mutasyonlar tip 1’de miktarsal azalmaya yol açarken, tip 2 konformasyon bozuklukları (kalitatif bozukluk) oluşturur, tip 3 ise neredeyse tümüyle VWF eksikliğine (şiddetli kantitatif bozukluk) neden olur. Tip 2 alt grupları (2A, 2B, 2M, 2N) fenotipik teste dayalı sınıflandırma ile ayırt edilir. Örneğin 2A’da yüksek moleküler ağırlıklı multimerlerin eksikliği vardır; 2B’de VWF’ün GPIb bağlanması artarak trombositlerde ve retiküloendoteliyal sistemde çökme ve trombositopeni gelişir; 2M’de fonksiyon bozukluğu olmasına rağmen multimer dağılımı normaldir; 2N’da ise VWF ile FVIII bağlanması bozulmuştur, bu nedenle FVIII düzeyi düşüktür (klinik olarak hafif hemofili A benzeri tablo).
Kalıtım çoğunlukla otozomal dominant olup, tip 1 ile 2A/2B/2M vakalarında baskındır (fenotip penetransı yüksek). Buna karşılık tip 2N ve tip 3 reseptif geçişlidir; tip 3 vakalarının çoğu homozigot veya kompoze heterozigot mutasyonlardan kaynaklanır. Tip 1’de penetrans tamamlanmamış olabilir; tip 3’te vakaların ~%80’i nonsense, delesyon veya çerçeve kayması gibi null alleller içerir. ABO kan grubu da VWF seviyelerini modifiye eder (0 grubu bireylerde daha düşük temel VWF düzeyleri gözlenir).
Epidemiyoloji:
VWD, tüm popülasyonda görece yaygındır ancak çoğu hafif seyreder. Orphanet’e göre dünya genelinde genel popülasyonun %0.6–1.3’ünde VWD mevcuttur; semptomatik olgularda prevalans yaklaşık 10/100.000’dir. Yüksek gelirli ülkelerde kayıtlı hasta prevalansı hemofili merkez verilerine göre ~1.5/100.000 civarındadır. Örneğin İngiltere Ulusal Veri Bankası’nda tüm tipleri kapsayan referral-prevalans 16.5/100.000 bulunmuştur (tip 1: 7.2, tip 2: 2.5, tip 3: 0.3/100.000). Tip 1 VWD en yaygın türdür (~vakaların %70–80’i), tip 2 %15–25, tip 3 ise nadir (%5’in altında) görülür. Tip 3 VWD’nin görülme sıklığı genel popülasyonda yaklaşık 0.5–1/1.000.000 olup, akraba evliliğinin yaygın olduğu topluluklarda bu değer 6/1.000.000’a kadar çıkabilir. Hastalar arasında cinsiyet dağılımı eşit olmakla birlikte, kadınlar adet kanaması, doğum ve gebelik gibi durumlar nedeniyle daha sık belirti bildirir. CDC verilerine göre belgelenen VWD olguları erkek ve kadınlarda yaklaşık eşit oranda olup, popülasyonda en fazla %1’e kadar kanama riskini artırabilir.
Klinik Görünüm:
VWD’nin temel semptomu mukokütan kanamadır. Bunlar arasında kolay çürük (purpura), küçük yaralardan uzun süren kanama, burun kanaması (epistaksis), diş eti kanamaları, aşırı adet kanaması (menorrhagi), gastrointestinal kanamalar ve cerrahi/diş çekimi/çocuk doğumu gibi hemostaz gerektiren durumlarda kanama bulunur. Hastalar arasındaki fenotip değişkenliği yüksektir; aynı ailedeki bireylerde bile şiddet hafif ila ciddi arasında değişebilir. Tip 1 VWD genellikle hafif veya orta kanama eğilimi gösterir ve bazı bireylerde sadece hemostatik zorlama ile ortaya çıkar (örneğin cerrahi sonrası). Tip 2’lerde kanama genellikle tip 1’den daha belirgindir; örneğin tip 2B’de trombositopeniye bağlı trombositleri uyaran anormal platelet bağlanması nedeniyle burun kanaması ve ekimoz sık görülebilir. Tip 2N’da ise düşük FVIII nedeniyle ekimoz ve travmatik kanamalar tipik olup, kadınlarda menorrhaji belirgindir. Tip 3 VWD en şiddetli klinik tabloyu verir; neredeyse hiç VWF bulunmadığı için spontan hematomlar, hemartrozlar (eklem içi kanamalar), ciddi post-operatif kanamalar ve ağır menstrüasyon görülür. Şiddetli tip 3 olgularında osteoartropati gelişebilmekte, eklem hasarına yol açabilmektedir. VWD’li kadınlarda menorrhagi ve doğum sonrası kanamalar oldukça yaygındır.
Tanı:
VWD tanısı klinik değerlendirme ile başlar. Özellikle aile öyküsü ve ISTH-BAT gibi kanama değerlendirme araçları kullanılır. Laboratuvar ilk bakışta kompleks bir inceleme gerektirir; tek bir test tanıyı doğrulamaz. Öncelikle tam kan sayımı (TKH) normaldir, ancak kronik kanamalarda anemi görülebilir. Koagülasyon testleri genellikle normaldir (PT, fibrinojen normal; APTT şiddetli olgularda uzayabilir). Spesifik tanı için ölçülen başlıca parametreler şunlardır: faktör VIII koagülan aktivitesi (FVIII:C), von Willebrand faktör antijeni (VWF:Ag), VWF işlevi ölçen testler (en yaygını VWF ristoseptin ko-faktör aktivitesi, VWF:RCo) ve VWF multimer dağılım analizi. VWF:Ag ve VWF:RCo seviyeleri düşüktür (tip 1’de her ikisi orantılı azalır; tip 2’de orantısızlık bulunabilir). Kolajen bağlama (VWF:CB) testi, RIPA (ristoseptin-indüklenen platelet aglütinasyonu) testi gibi ek fonksiyonel ölçümler de kullanılır. Tanı şeması genellikle VWF:Ag, VWF aktivite, FVIII ve multimer analizini içerir. Örneğin, VWF:Ag ile VWF aktivitelerinden herhangi biri <%30 (0.30 IU/mL) ise VWD düşünülür; %30–50 düzeylerinde ise kanama semptomlarının eşlik etmesi tanıyı destekler. Ayırıcı tanıda hemofili A/B, Bernard-Soulier sendromu, primer platelet fonksiyon bozuklukları, antiplatelet ilaç kullanımı ve “psödo”/platelet-tipi VWD göz önünde bulundurulmalıdır. Genetik test özellikle tip 2B, 2N ve tip 3’ün ayırıcı tanısında yararlıdır. Tip 2N vakalarında VWF genindeki ilgili mutasyonların bulunması hemofili A ile karışmayı önler. Tip 1’lerde ise genetik test nadiren yardımcı olur (polimorfizm etkileri, penetrans değişkenliği çoktur).
Tedavi:
VWD tedavisi kanamayı durdurmaya ve önlemeye yöneliktir. Başlıca hemostatik ajanlar şunlardır: Desmopressin (DDAVP) ve VWF içeren faktör konsantreleri. Desmopressin, endojen VWF ve FVIII depolarından salgılatır; tip 1 ve çoğu tip 2 (2A, 2M) olgularında ilk seçenek tedavidir. DDAVP kolay uygulanması, maliyetinin düşüklüğü ve ek kan ürünü kullanımını azaltması nedeniyle tercih edilir. Ancak tip 2B’de trombositopeniyi kötüleştirebileceğinden dikkatli kullanılmalı, tip 3’te ise yeterli depolanan VWF olmadığından etkisizdir. Major kanamalarda veya cerrahi için VWF/FVIII içeren plazma kaynaklı konsantreler (VWF içerikli faktör VIII preparatları) kullanılır. Rekombinant VWF preparatları (FVIII içermeyen) da mevcuttur. Antifibrinolitik ajanlar (traneksamik asit, aminokaproik asit) mukozal kanamalar ve menorrhaji tedavisine destek olarak sıkça eklenir. Kadınlarda aşırı adet kanamalarında hormon replasmanı veya intrauterin sistem yöntemleri yardımcı olabilir. Kanama atakları dışındaki önleyici (profilaktik) tedavi, tekrar eden eklem veya ciddi kanamalarda düşünülür; özellikle tip 3 hastalarında düzenli VWF konsantresi infüzyonları hem birikimli eklem hasarını hem de kanama komplikasyonlarını azaltır. Ayrıca, cerrahi, dental çekim, doğum gibi invaziv girişimler öncesinde hemostatik profilaksi sağlamak için DDAVP veya VWF konsantreleri uygulanır. Tüm VWD hastaları, kanama semptomları ciddiye dönerse bir hematoloji merkezinde değerlendirilmelidir.
Keşif
Von Willebrand hastalığı (VWD), ilk kez 1926 yılında, Finli doktor Erik Adolf von Willebrand tarafından tanımlanmıştır. Bu hastalık, genetik bir kanama bozukluğudur ve ilk olarak, şiddetli kanamalarla tanı almış bir kadın hastanın vakası üzerinde yapılan araştırmalarla ortaya çıkmıştır. Von Willebrand, hastalığın, “kanama eğilimi” ile karakterize olduğunu ve özel bir kanama sendromu oluşturduğunu belirtmiştir. Bu araştırma, genetik ve klinik faktörlerin kanama bozuklukları üzerindeki etkisini ilk defa açıklığa kavuşturmuştur.
Erik von Willebrand, 1926 yılında yazdığı makalesinde, hastalığın Finlandiya’nın çeşitli bölgelerinde yaygın olduğuna dikkat çekmiş ve hemofili A ve B hastalıklarından farklı olduğunu belirtmiştir. Bu hastalığın, damarların iç yapısındaki bazı özel proteinlerin eksikliği veya fonksiyon bozukluğu ile ilişkili olduğunu ilk kez ileri sürmüştür. Bu durumu daha iyi anlamak için von Willebrand, hastaların kanama geçmişini inceledi ve damarların pıhtılaşma süreçlerinde önemli bir rol oynayan faktörleri tespit etmeye başladı.

Sonraki yıllarda, 1950’ler ve 1960’larda von Willebrand hastalığının patofizyolojisi ve genetik temelleri daha ayrıntılı bir şekilde incelenmeye başlandı. Ancak, hastalığın temel etiyolojik nedenleri ve biyokimyasal yönleri hakkında tam bir anlayış, 1970’ler ve 1980’ler boyunca genetik araştırmaların ve koagülasyon faktörlerinin daha iyi tanımlanması ile mümkün olabilmiştir.
Faktör VIII ve Von Willebrand Faktörü: Genetik ve Klinik Yansıması
Hastalığın moleküler temeli, 1950’lerde yapılan çalışmalarla hızla ilerlemeye başlamıştır. Bu dönemde, von Willebrand faktörünün (VWF) önemli bir koagülasyon faktörü olduğu anlaşılmıştır. VWF, faktör VIII’i taşıyarak onu stabilize eder ve aynı zamanda damar hasarı sonrası trombositlerin yapışmasını teşvik eder. Bunun yanı sıra, VWF’nin büyük boyutlu multimerik yapılarla kanama sırasında işlev gördüğü de belirlenmiştir.
Faktör VIII’in stabilitesi ve işlevi, VWF’ye bağımlıdır. Von Willebrand hastalığının ilk tanımları bu iki faktör arasındaki ilişkilerin anlaşılmasında önemli bir dönüm noktası yaratmıştır. 1970’lerde, faktör VIII ve von Willebrand faktörünün eksikliklerinin kanama bozukluklarına neden olduğu anlaşılmış, bu iki faktör arasındaki işlevsel ilişki moleküler düzeyde açıklığa kavuşmuştur.
Von Willebrand Hastalığının Genetik Yönü:
Von Willebrand hastalığının genetik temeli üzerine yapılan çalışmalar, 1980’lerde büyük bir hız kazandı. Von Willebrand faktörünün genetik yapısı, kromozom 12’de yer alan bir genin etkisiyle ortaya çıkar. Bu genetik yapı üzerinde yapılan araştırmalar, hastalığın kalıtım tarzlarını (otozomal dominant ve otozomal resesif) ve mutasyon türlerini ortaya koymuştur. Bu çalışmalar, hastalığın daha iyi anlaşılmasını sağlamış ve tanısal testlerin gelişmesine katkı sunmuştur.
Sonraki Keşifler:
1990’lar, von Willebrand hastalığının daha ayrıntılı genetik incelemelerine sahne olmuştur. VWF genindeki çeşitli mutasyonlar belirlenmiş, bu mutasyonların hastalığın şiddetini ve klinik seyrini nasıl etkilediği açıklığa kavuşmuştur. Ayrıca, hastalığın tipleri (tip 1, tip 2 ve tip 3) arasındaki farklar, daha hassas laboratuvar testleri ile netleşmiş ve klinik tanı daha güvenilir hale gelmiştir.
Genetik Bilgiler ve Tanı Yöntemleri:
Von Willebrand hastalığının tanısal aşamaları, özellikle genetik analizlerin gelişmesiyle önemli bir evrim geçirmiştir. 1990’ların sonlarına doğru, genetik testler ve mutasyon analizleri, hastalığın doğru şekilde sınıflandırılmasında önemli bir araç haline gelmiştir. Sonuç olarak, von Willebrand hastalığı, hemofili ile karışabilecek şekilde klinik bir tabloya sahipken, yapılan laboratuvar testleri sayesinde doğru şekilde tanımlanabilmektedir.
Sonuç olarak: Von Willebrand hastalığının keşfi ve patofizyolojik temellerinin anlaşılması, birkaç on yıl süren bir süreçtir. İlk tanımlamadan günümüze kadar olan süreçte, hastalığın moleküler biyolojisi, genetik temelleri ve klinik özellikleri hakkındaki anlayışımız büyük bir gelişim göstermiştir. Hem genetik hem de biyokimyasal düzeydeki ilerlemeler, tanı ve tedavi yaklaşımlarının daha hassas ve etkili hale gelmesini sağlamıştır.
İleri Okuma
- Cumming AM, Keeney S, Jenkins PV, Nash MJ, O’Donnell JS. Clinical utility gene card for: von Willebrand disease. Eur J Hum Genet. 2011;19(5).
- Roberts JC, Flood VH. Laboratory diagnosis of von Willebrand disease. Int J Lab Hematol. 2015;37 Suppl 1:11–17.
- Du P, Bergamasco A, Moride Y, et al. Von Willebrand Disease Epidemiology, Burden of Illness and Management: A Systematic Review. J Blood Med. 2023;14:189–208.
- James PD, Connell NT, Ameer B, et al. ASH ISTH NHF WFH 2021 guidelines on the diagnosis of von Willebrand disease. Blood Adv. 2021;5(1):280–300.
- Beltran A, Jaramillo AP, Vallejo MP, Acosta L, Parraga GCB. Desmopressin as a treatment in patients with von Willebrand disease: A systematic review. Cureus. 2023;15(8):e44310.
- GeneReviews®: Johnsen J. von Willebrand Disease. 1993–2024 (güncellenmiş 14 Kas 2024). Seattle: University of Washington; (erişim: NCBI Bookshelf).