Kronik miyeloid lösemi ya da kısaca KML, kemik iliğindeki hematopoetik kök hücrelerin kötü huylu klonal bir hastalığıdır.
ICD-10 kodu
C92.1 Kronik miyeloid lösemi, BCR/ABL-pozitif
C92.2 Atipik KML, BCR/ABL negatif
C94.8 KML’de patlama krizi
Sınıflandırma
Klasik KML: Klasik KML miyeloproliferatif hastalıklardan biridir. Moleküler patolojik temeli BCR-ABL1 translokasyonudur.
Atipik BCR-ABL1-negatif KML: Atipik KML, miyelodisplastik-miyeloproliferatif neoplazmlar (MDS/MPN) grubuna aittir.
Ana maddeye bakınız: Atipik kronik miyeloid lösemi
Epidemiyoloji
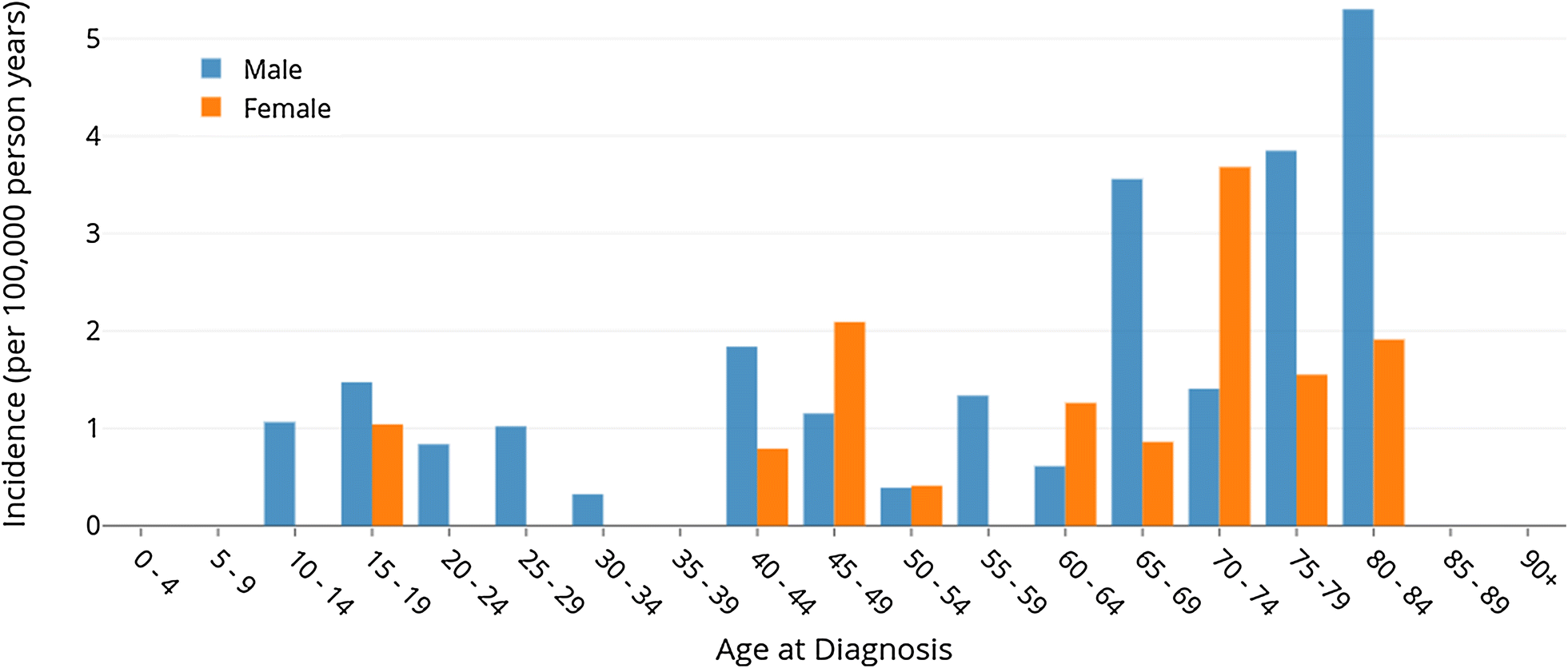
KML insidansı yılda 100.000 kişi başına yaklaşık 1,2 – 1,5’tir. Her yıl yaklaşık 1.000 – 1.500 kişi hastalanmaktadır. Hastaların %60’ı erkektir.
Yetişkinlerdeki lösemi vakalarının dörtte biri kronik miyeloiddir. KML tüm yaş gruplarında görülmekle birlikte, hastalık 55 – 65 yaşlarında zirve yapar. KML çocuklarda çok nadir görülür. KML hastalarının sadece %3’ü 20 yaşın altındadır.
Etiyoloji
Kronik miyeloid löseminin kesin nedeni bilinmemektedir. Kemik iliğinin pluripotent kök hücrelerinde kötü huylu bir dejenerasyon vardır. Ailesel bir küme yoktur ve toksinlere, böcek ilaçlarına veya virüslere maruz kalma ile bir ilişki olduğuna dair net bir kanıt yoktur. Bununla birlikte, benzene kronik maruziyet, belirli koşullar altında bir meslek hastalığı olarak kabul edilebilir.
KML sadece nadiren kemoterapi veya radyoterapiye bağlı olarak ortaya çıkar. İyonlaştırıcı radyasyon, maruziyetten en fazla 5 ila 10 yıl sonra olmak üzere doza bağlı bir şekilde riski artırır. Hiroşima’ya atılan atom bombasından kurtulanlarda KML’nin ortaya çıkmasına kadar geçen medyan süre 6,3 yıldı. Ancak, Çernobil nükleer felaketinden sonra insidans artmamıştır; muhtemelen sadece yüksek dozlar KML’ye yol açmaktadır.
Patogenez
Translokasyon
KML hastalarının %95’inden fazlasında spesifik bir kromozomal aberasyon olan dengeli resiprokal translokasyon t(9;22)(q34;q11) bulunur. Özellikle kısa bir kromozom 22’ye, Philadelphia kromozomuna (Ph) yol açar.
Bu süreçte, kromozom 9’daki protoonkogen ABL1’in (Abelson tirozin kinaz) DNA dizileri, kromozom 22’deki kırılma noktası küme bölgesi geninin (BCR) bölgesine yerleştirilir. BCR-ABL füzyon geni artık yapısal olarak aktif bir tirozin kinaz olan onkojenik füzyon proteini BCR-ABL1’i kodlamaktadır. Bu da KML hücrelerinin aşırı çoğalmasına ve apoptozun azalmasına neden olur. Hastalığın seyri sırasında fizyolojik hematopoez baskılanır. Bununla birlikte, normal kök hücreler etkili bir tedaviden sonra devam edebilir ve tekrar aktif hale gelebilir.
Translokasyon miyeloid hücrelerde (eritrositik hücreler, megakaryositler ve monositler dahil), daha az sıklıkla olgun B ve T hücrelerinde tespit edilebilir, ancak kemik iliğinin stromal hücrelerinde veya diğer somatik hücrelerde tespit edilemez.
BCR-ABL1 translokasyonu ile KML gelişimi arasında nedensel bir ilişki olduğu deneysel modellerde kanıtlanmıştır. Bununla birlikte, BCR-ABL1 transkriptleri sağlıklı bireylerde de bulunabilir. Bu da BCR-ABL’nin tek başına KML’yi tetiklemediği sonucuna götürmektedir. Muhtemelen, daha fazla moleküler olay veya BCR-ABL’li hücrelerin hatalı bağışıklık sistemi tarafından tanınması gereklidir.
Varyantlar
Füzyon geninin terapötik önemi de olan çeşitli mutasyonları mevcuttur. Moleküler ağırlığa bağlı olarak, üç tip füzyon proteini ayırt edilebilir. P210 en yaygın formdur ve e13a2 ve e14a2 transkriptleri tarafından kodlanır.
Daha kısa bir BCR dizisi ABL1 ile birleştiğinde, daha küçük BCR-ABL1 onkoproteini p190 (transkript e1a2) oluşur. KML’de nadiren bulunur ve daha sonra daha kötü bir prognozla ilişkilendirilir. Ancak P190, Ph-pozitif akut lenfoblastik lösemili (ALL) hastaların üçte ikisinde bulunur.
Üçüncü form p230, e19a2 transkripti tarafından kodlanır ve indolent bir seyir ile ilişkilidir.
Seyir
Belirli tedavi biçimlerinin kullanılmasından önce, çoğu hasta tipik olarak üç aşamalı bir seyir göstermekteydi. Yaygın semptomlar özellikle sağlık hizmetlerinin mevcudiyetine ve bu hizmetlere erişime bağlıdır.
Almanya’da KML, rutin kan testleri sırasında hastaların %50-60’ında tesadüfen teşhis edilmektedir. Sağlık hizmetlerinin yetersiz olduğu ülkelerde KML, semptomlar veya komplikasyonlar ortaya çıktığında geç teşhis edilmektedir.
Bireysel evreler DSÖ’nün 2016 tanımına göre veya Avrupa Lösemi Ağı (ELN) kriterleri kullanılarak tanımlanabilir.
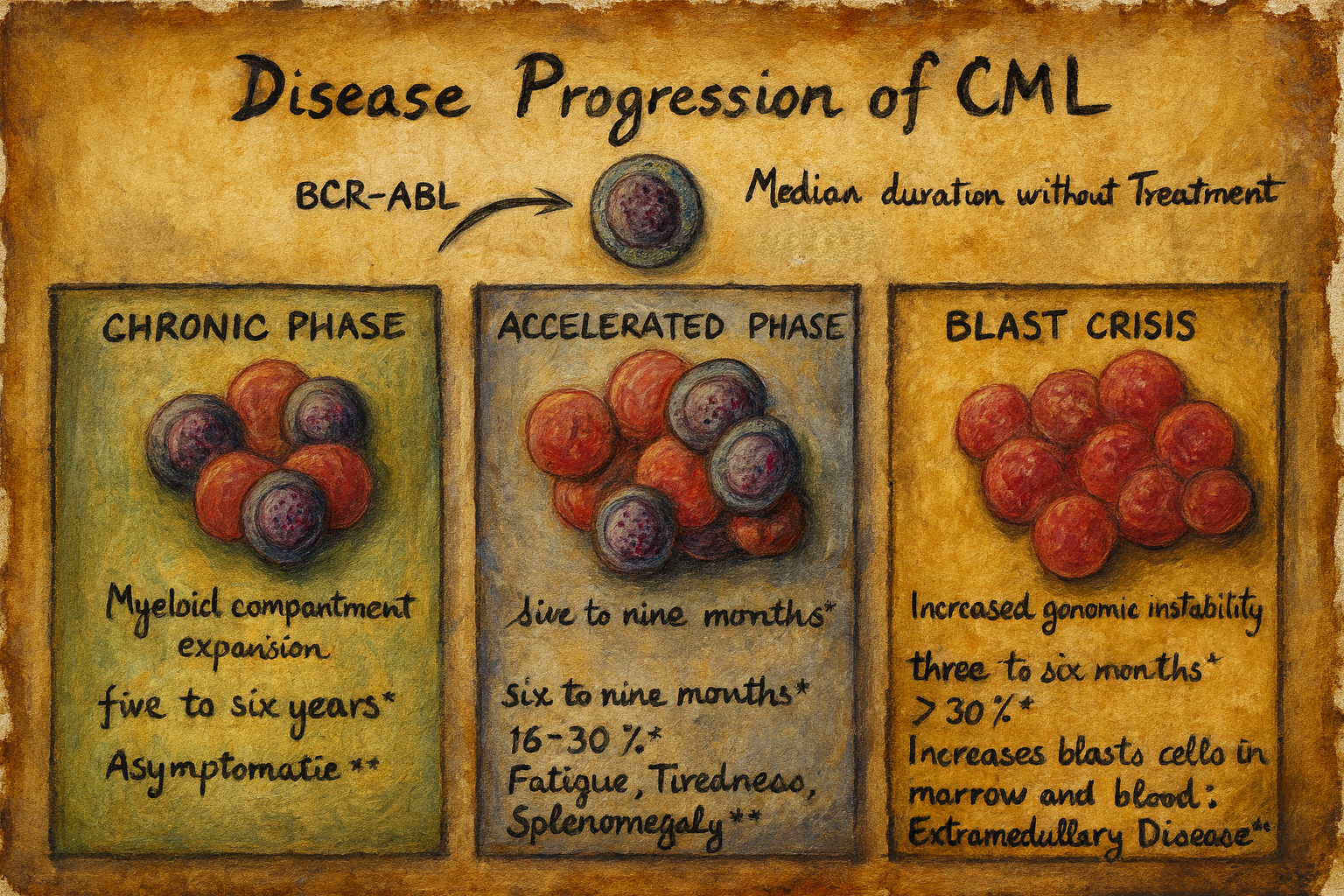
Kronik stabil faz
KML kronik stabil faz (KP) ile başlar. Bu durum 6 aydan 20 yıla kadar sürer. Hastaların %90 ila 95’i ilk tanıda SP’dedir.
Tanım olarak, periferik kanda ≤ %2 ve kemik iliğinde ≤ %5 blast vardır.
Hızlanma aşaması
Hızlanma fazı (AP), kronik faz ile blast nüksü arasındaki geçişi ifade eder. Bu muhtemelen başka kromozomal değişikliklerden kaynaklanmaktadır. ELN tanımına göre, aşağıdaki kriterlerden en az birinin mevcut olması halinde bir hızlanma aşaması mevcuttur:
- 15 – 29 Kan veya kemik iliğinde % blast
- Kan veya kemik iliğinde %30’dan fazla blast ve promyelosit (< %30 blast ile) Kan veya kemik iliğinde en az %20 bazofil Tedaviden bağımsız trombositopeni < 100.000/µl Trombositoz > 1.000.000/µl
- yeni oluşan klonal evrim
- ilerleyici kemik iliği fibrozu
- refrakter progresif splenomegali ve artan lökosit sayısı
DSÖ, hızlanma evresini aşağıdaki kriterlerden en az birinin varlığı olarak tanımlamaktadır:
- Tedaviye rağmen kalıcı veya artan lökositoz > 10.000/µl
- tedaviye rağmen devam eden veya artan splenomegali
- Tedaviye rağmen kalıcı trombositoz > 1.000.000/µl
- Kalıcı trombositopeni < 100.000/µl, tedaviden bağımsız Periferik kanda > %20 bazofil
- Periferik kan ve/veya kemik iliğinde %10 – 19 blast
- Philadelphia kromozomu pozitif hücrelerde ek sitogenetik aberasyonlar (örn. izokromozom 17)
- tedavi altındaki Philadelphia kromozomu pozitif bir hücrede herhangi bir yeni klonal kromozomal sapma
- ilk tirozin kinaz inhibitörüne (TKI) karşı hematolojik direnç veya ilk TKI’ye tam hematolojik yanıt alınamaması
- Ardışık olarak uygulanan iki TKI’ye karşı direnç olduğuna dair herhangi bir hematolojik, sitogenetik veya moleküler kanıt.
- Tedavi sırasında iki veya daha fazla BCR-ABL1 mutasyonunun ortaya çıkması.
Patlama krizi
Tedavi olmaksızın, blast krizi tipik olarak ilk tanıdan 3 ila 5 yıl sonra ortaya çıkar. DSÖ tanımına göre kanda veya kemik iliğinde %20’den fazla, ELN sınıflandırmasına göre ise %30’dan fazla blast bulunmaktadır.
Hem WHO hem de ELN’ye göre bir diğer tanı kriteri de ekstramedüller blastların çoğaldığının kanıtlanmasıdır (örneğin ciltte, yumuşak dokuda veya osteoliz şeklinde).
Blast krizinin seyri akut lösemiye benzer ve tedavi edilmezse hızla ölümle sonuçlanır.
Vakaların üçte ikisi myeloid (nadiren eritrositik, promyeloid, monositik, megakaryositik), üçte biri lenfatik blast krizleridir. Lenfoid blast krizi, bir TKI ile birlikte ALL protokolüne (siklofosfamid, vinkristin, doksorubisin, deksametazon) göre kemoterapiye yanıt verebileceğinden, bu ayrımın terapötik bir sonucu vardır.
Klinik tablo
Başlangıçta, kronik stabil fazdaki hastaların %50’si asemptomatiktir. Hastalık ne kadar ilerlerse belirtiler de o kadar erken ortaya çıkar. KML’nin önde gelen semptomu sürekli sola kayan lökositozdur. Diğer semptomlar veya bulgular şunlardır:
Spesifik olmayan genel semptomlar: Genel hastalık hissi, B semptomları (kilo kaybı, ateş, gece terlemesi), yorgunluk, performans düşüklüğü.
Splenomegali: Kırmızı dalak pulpasının olgun ve olgunlaşmamış granülositler tarafından infiltrasyonundan kaynaklanır; sol üst karın bölgesinde basınç hissine ve erken dolgunluk hissine yol açabilir.
Anemi
- Trombositoz veya daha nadiren trombositopeni.
- Bazofili: esas olarak hızlanma aşamasına geçişi gösterir
- Göğüs kafesi bölgesinde vurma veya basınç ağrısı
- Aşırı üretilen granülositler işlevsel olduğundan, akut löseminin aksine başlangıçta tekrarlayan enfeksiyonlar ortaya çıkmaz. Hızlanma evresinde, daha düşük farklılaşma kapasitesine sahip olgunlaşmamış BCR-ABL-pozitif blastlardaki artış nedeniyle giderek daha az sayıda işlevsel olgun granülosit bulunur. Bu da enfeksiyon riskini artırır.
Komplikasyon
Komplikasyonlar, periferik kan hücrelerinin sayısının veya işlevinin değişmesinden kaynaklanır. Trombositoz tromboz riskinde artışa, trombopeni ise kanama eğiliminde artışa neden olur. Buna ek olarak, von Willebrand faktörünün trombositlere bağlanması sonucu edinilmiş von Willebrand sendromu ortaya çıkabilir.
Granülositler ve öncülleri kolayca deforme olabildiğinden, reolojik sorunlar nadiren ortaya çıkar. Bununla birlikte, aşırı lökositoz vakalarında lösemik trombüsler mümkündür. Karşılık gelen belirtiler şunlardır:
- Splenik enfarktüs
- merkezi retinal ven trombozu
- lösemik priapizm
- Miyokard enfarktüsü
- pulmoner arter embolisi
- İnme
Teşhis
Tıbbi geçmiş
Öykü, yukarıdaki semptomlarla ilgili soruları içermelidir, örneğin yorgunluk, halsizlik, iştahsızlık, kilo kaybı, kemik ağrısı, üst karın ağrısı.
Fiziksel muayene
KML’de en sık görülen fiziksel bulgu, hastaların %20-70’inde bulunan splenomegalidir. Vakaların %10-20’sinde hepatomegali bulunur. Nadiren, ekstramedüller hematopoezin bir sonucu olarak blast içeren deri veya deri altı lezyonları gelişir. İkincisi, bir patlama krizini tanımlayan bir kriterdir. Lenfadenopati oldukça atipiktir ve vakaların yalnızca %5-10’unda görülür.
Diğer fiziksel bulgular bahsedilen komplikasyonlardan kaynaklanır, örneğin santral ven trombozunda görme bozuklukları. Yüksek bazofili, kaşıntı, ishal ve ateş basması ile birlikte aşırı histamin üretimine yol açabilir.
Kan testi
KML’den şüpheleniliyorsa, diferansiyel kan sayımı istenmelidir. Aşağıdaki bulgular öne çıkmaktadır:
- Anemi: Hastaların yaklaşık %33-80’inde çoğunlukla hafif bir anemi görülür.
- Özellikle nötrofil granülositlerindeki artışa bağlı lökositoz: Diğer lösemi türlerinin aksine, KML’de lökositlerde her zaman önemli bir artış vardır. Hatta KML tüm lösemiler arasında en yüksek lökosit sayısına sahiptir, bazen 500.000/µl’nin üzerinde değerler görülebilir. Yüksek lökosit sayıları, kan düşürüldüğünde geniş bir lökosit manşeti şeklinde zaten fark edilebilir.
- Sola kayma: Nötrofillerin baskın olduğu ve miyeloblastlara kadar tüm olgunlaşma aşamalarının bulunduğu lökositler bulunur (lösemoid reaksiyon veya akut löseminin aksine).
- Bazofili, eozinofili: Bazofilik ve eozinofilik granülositlerde artış sıklıkla gözlenir.
- Özellikle trombosit sayısında artış (trombositoz) vakaların %50’sinde, trombosit sayısında azalma (trombositopeni) vakaların %10’unda görülür ve genellikle trombositlerin fonksiyonel bir bozukluğu (trombositopati) da mevcuttur.
- Seyir sırasında, normal hematopoezin yer değiştirmesi ile miyelofibroz gelişir. Sonuç olarak, örneğin dalakta ekstramedüller hematopoez meydana gelir. Çekirdekli kırmızı öncüller periferik kanda görülebilir.
Ayrıca, B12 vitamini, ürik asit, LDH ve lizozim plazma konsantrasyonlarında artış tespit edilmiştir. ESR tipik olarak yükselmiştir.
BCR-ABL transkriptlerinin sitogenetik tespiti PCR ile gerçekleştirilir.
Kemik iliği incelemesi
Kemik iliği aspiratında sitolojik, sitogenetik ve sitokimyasal incelemeler yapılabilir:
- Sitoloji: hipersellülarite, baskın granülopoez (G-E indeksi artmış), küçük megakaryositler, yalancı Gaucher hücreleri veya deniz mavisi histiyositler, bazofili, eozinofili, genellikle %5’ten az blast (kronik faz).
- Sitogenetik: BCR-ABL füzyon geninin veya Philadelphia kromozomunun saptanması ile metafaz analizi.
- Sitokimya: Alkalin lökosit fosfataz (ALP) aktivitesi güçlü bir şekilde azalmıştır. Bu, KML’yi diğer tüm miyeloproliferatif hastalıklardan ayırır.
- Kemik iliği biyopsisi histolojik olarak incelenir. Fibrozise ek olarak promyelosit ve myelositlerde artış vardır.
Sitogenetik-moleküler biyolojik inceleme
Klasik KML’de Philadelphia kromozomu ve BCR-ABL füzyon geni FISH veya RT-PCR ile tespit edilebilir. Philadelphia kromozomu veya BCR-ABL transkriptlerinin saptanması, böylece tanıyı güvence altına alır. Kan ve kemik iliğinde BCR-ABL miktarının ölçülmesi de tedavinin izlenmesine hizmet eder.
Bazı hastalarda ek kromozomların yer değiştirdiği kompleks translokasyonlar (varyant Ph kromozomu) vardır. Diğer hastalarda kromozom 9 ile kromozom 22 dışında bir kromozom arasında translokasyon ile maskelenmiş bir Ph kromozomu vardır. Bu vakalarda prognoz ve tedavi klasik KML ile karşılaştırılabilir.
Buna ek olarak, Ph-pozitif hücrelerin %5 ila 10’unda başka aberasyonlar (örn. trizomi 8, izokromozom 18, 17p delesyonu) bulunur.
İmmünofenotipleme
İmmünofenotipleme, özellikle blast krizi sırasında blastları karakterize etmek için kullanılır. Bunun tedaviye yanıt ve prognoz açısından önemi vardır.
Ayırıcı tanılar
- Miyeloid lösemoid reaksiyon: Kronik pürülan enfeksiyonlar, sepsis veya G-CSF uygulaması gibi durumlarda lökositoz görülebilir. Lökosit sayısı genellikle 100.000/µl’nin altında kalır. Periferik kanda, toksik granülasyon olarak adlandırılan granülositler vardır, ancak bazofili yoktur ve çok nadiren miyeloblastlar bulunur.
- Lenfositik lösemoid reaksiyon: Bazı viral enfeksiyonlarda veya boğmacada yüksek lenfosit sayıları görülür.
- Kronik miyelomonositik lösemi: Ph-negatif, tip I ve II’de ALP yüksekliği
- Diğer miyeloproliferatif hastalıklar, örneğin kronik nötrofil lösemi (CSF3R mutasyonu)
- Primer miyelofibrozis: splenomegali, sola kayan lökositoz, trombositoz, ALP artışı
- Atipik KML: CSF3R veya SETBP1 mutasyonları, displastik granülositopoez (hiposegmente psödo-Pelger hücreleri)
- Miyelodisplastik sendrom (MDS): MDS’nin aksine, KML’deki periferik kan hücreleri çok az displazi belirtisi gösterir.
- ALL: Yetişkin hastaların %20’sinde ve çocuk hastaların %5’inde Philadelphia kromozomu bulunur.
- Akut miyeloid lösemi (AML): Tüm hastaların %2’sinden azında Philadelphia kromozomu vardır. AML’de, periferik kandaki ara madde eksikliği nedeniyle hiatus leucaemicus olarak adlandırılan durum mevcuttur.
Terapi
Ana maddeye bakınız: Kronik miyeloid lösemi tedavisi.
Kronik evre
Tanı doğrulandıktan sonra tedavi, kontrollü klinik çalışmalara katılımın sağlandığı hematoloji merkezlerinde gerçekleştirilmelidir. Tedavinin amacı, kan sayımının remisyon anlamında kapsamlı bir şekilde normalleştirilmesidir. Üç farklı remisyon şekli ayırt edilir:
- Hematolojik remisyon (HR)
- Tam hematolojik remisyon (CHR)
- İyi hematolojik remisyon (MHR, majör hematolojik remisyon)
- Sitogenetik remisyon (CyR)
- Tam sitogenetik remisyon (CCyR)
- İyi sitogenetik remisyon (MCyR, majör sitogenetik remisyon)
- Moleküler remisyon (MR)
- Tam moleküler remisyon (CMR)
- İyi moleküler remisyon (MMR, majör moleküler remisyon)
- Tirozin kinaz inhibitörleri ilk basamak tedavi için önerilmektedir. Yaş, cinsiyet, komorbiditeler ve olası yan etkilere bağlı olarak imatinib, nilotinib, dasatinib veya bosutinib kullanılır. TKI’ler yaklaşık %60 ila 80 oranında moleküler remisyona yol açmaktadır. Tedavi ile 10 yıllık sağkalım oranı %80 ila 90’dır.
Tedaviye yanıt için tanımlanmış kriterler vardır: Optimal durumda, yaşam beklentisi neredeyse normaldir; tedaviye yanıt alınamaması durumunda, mutasyon analizi ve ikinci basamak tedavi hızlı bir şekilde yapılmalıdır. Daha önce kullanılmamış bir TKI veya ponatinib düşünülebilir.
Prensip olarak, allojenik kök hücre transplantasyonu (SCT) her aşamada potansiyel olarak iyileştirici bir tedavi olarak düşünülmelidir. Allojeneik SCT sonrası 10 yıllık sağkalım oranı yaklaşık %55’tir.
Hızlanma aşaması
Hızlanma evresinde olan ve daha önce bir TKI ile ön tedavi görmüş hastalarda, mutasyon durumuna bağlı olarak alternatif bir TKI kullanılır.
Ön tedavisi olmayan hastalarda imatinib, bosutinib, dasatinib ve nilotinib ve uygun hastalar ve donör mevcudiyeti varsa allojenik kök hücre nakli de düşünülmektedir.
Patlama krizi
Miyeloid blast krizinde hidroksiüre, sitarabin ve muhtemelen antrasiklinler yardımcı olurken, lenfoid formda diğerlerinin yanı sıra vinkristin ve deksametazon kullanılır.
Ayrıca, hızlandırma aşamasında olduğu gibi, tüm TKI’ler (nilotinib hariç) ve allojenik SCT düşünülebilir.
Prognoz
TKI’lerin kullanılmaya başlanmasından önce hidroksiüre, busulfan ve interferon-alfa tedavisi altında ortalama sağkalım süresi 3 ila 7 yıl, 10 yıllık sağkalım oranı ise %30 idi. İlk iki yılda yıllık ölüm oranı %10, daha sonra %15 ila %20 olmuştur.
Bu arada, allojenik SCT tedaviye yol açabilirken, TKI’ler CMR varlığında bile muhtemelen küratif değildir, çünkü çok erken lösemik kök hücreler ortadan kaldırılmaz. Bununla birlikte, TKI’ler genellikle neredeyse normal bir yaşam beklentisine yol açar. Bu anlamda KML indolent bir hastalık olarak kabul edilebilir.
Prognozun klinik değerlendirmesi için üç skor mevcuttur.
- Sokal ve Hasford’a göre EURO skoru: TKI tedavisinin başlamasından önceki verilere dayanmaktadır
- EUTOS skoru: CCyR’ye ulaşılmasını öngörmek için periferik kandaki bazofil oranını ve tanı anında dalak boyutunu kullanır
- ELTS skoru: şu anda (2019) önerilen prognoz skoru
Keşif
Kronik Miyeloid Lösemi (KML) keşfinin tarihi, 180 yılı aşkın bir süreyi kapsayan, önemli klinik gözlemler, genetik atılımlar ve terapötik ilerlemelerle işaretlenmiş, büyüleyici bir tıbbi ve bilimsel ilerleme yolculuğudur.
Erken Klinik Tanıma (1840’lar-1870’ler)
KML’nin tarihi, Fransa, Almanya ve İskoçya’da görülen erken vakalarla 1840’larda başlar. 1841’de Glasgow’da David Craigie, daha sonraki raporlarda açıklandığı gibi, muhtemelen KML olan ateş, splenomegali ve lökositozlu bir hastayı gözlemledi. Kesin tanıma, John Hughes Bennett’in Edinburgh Tıbbi ve Cerrahi Dergisi‘nde bir otopsi yapıp ayrıntılı bulgular sunarak KML’yi ilk lösemi türü olarak tanımlamasıyla 1845’te geldi. Aynı zamanda, Rudolf Virchow benzer vakaları tanımladı ve anormal beyaz kan hücresi çoğalmasını vurgulayan “Leukämie” terimini ortaya attı. Ernst Neumann’ın 1871 tarihli çalışması löseminin kemik iliğinden kaynaklandığını daha da açıklığa kavuşturdu ve hematolojik doğasını anlamak için temel oluşturdu.
19. yüzyılda tedavi ilkeldi ve arsenik, lökosit sayısını azaltan ancak hayatta kalma açısından çok az fayda sağlayan tek iyi belgelenmiş tedaviydi. Arthur Conan Doyle’un 1882 tarihli Lancet raporu, dönemin sınırlı seçeneklerini yansıtan arsenik kullanımını vurguladı.
Genetik Keşifler: Philadelphia Kromozomu ve Ötesi (1960-1973)
20. yüzyıl dönüştürücü genetik içgörüler getirdi. 1960 yılında, Pennsylvania Üniversitesi’nden Peter Nowell ve David Hungerford, KML hastalarının beyaz kan hücrelerinde ve kemik iliğinde anormal bir “dakika kromozomu” tespit ettiler ve daha sonra konumlarına atfen Philadelphia (Ph) kromozomu adını verdiler. Bu, belirli bir kanserle bağlantılı ilk tutarlı kromozomal anormallikti ve Science dergisinde yayınlandı. Bu keşif, KML araştırmalarını genetiğe doğru kaydırdı ve Ph kromozomu hastalığın ayırt edici özelliği haline geldi.
1973 yılında, Janet Rowley, gelişmiş sitogenetik teknikler kullanarak, Ph kromozomunun 9 ve 22 numaralı kromozomlar arasındaki karşılıklı bir translokasyondan kaynaklandığını belirledi ve t(9;22) olarak adlandırıldı ve Nature dergisinde yayınlandı. Bu, kansere neden olan bir kromozomal translokasyonun ilk gösterimiydi ve bu bulgu kanser genetiğini kökten değiştirdi ve KML’nin farklı kimliğini sağlamlaştırdı.
Moleküler Biyoloji: BCR-ABL1’in Çözülmesi (1980’ler-1990’lar)
1980’ler, KML’nin moleküler temeline daha derin bir dalışın yapıldığı yıllardı. 1983’te Nora Heisterkamp ve Jim Groffen, çalışmalarında ayrıntılı olarak açıklandığı gibi, KML’de 22. kromozoma taşınan ABL1 genini (v-Abl’nin insan homologu) 9. kromozomda buldular. 1984’te Groffen ve arkadaşları, 22. kromozomda “kırılma noktası küme bölgesi”ni (BCR) tanımladılar ve tüm translokasyon kırılma noktalarının 5,8 kb’lik bir bölgede meydana geldiğini göstererek, KML’nin patogenezini anlamak için kritik bir bulgu olan BCR-ABL1 füzyon genine yol açtılar. 1985’te Shtivelman ve arkadaşları ve Stam ve arkadaşları, KML hastalarında bu füzyondan kimerik mRNA’yı tespit ederek moleküler mekanizmayı doğruladılar.
Ayrıca, 1984’te Konopka ve arkadaşları. Ph-pozitif K562 hücrelerinde yapısal tirozin kinaz aktivitesine sahip değişmiş bir c-ABL proteini (p210) gözlemlendi ve ilgili çalışmalarda yayınlandı. 1986’da Ben-Neriah ve arkadaşları, p210’un BCR/ABL hibrit geninin ürünü olduğunu doğruladı ve füzyonu kontrolsüz hücre büyümesine bağladı. Onkojenik potansiyel, 1990’da farelerde retroviral fare kemik iliği transdüksiyonu kullanarak CML benzeri bir bozukluk oluşturan Daley ve arkadaşları tarafından kanıtlandı ve araştırmalarında bildirildiği gibi BCR-ABL1’in hastalığa neden olma yeterliliğini gösterdi.
Tedavi Evrimi: Arsenikten Hedefli Terapilere
19. yüzyıldaki erken tedaviler, tarihsel incelemelerde belirtildiği gibi sınırlı etkililiğe sahip arseniklere dayanıyordu. 20. yüzyılın ilk yarısında radyoterapi görüldü, ardından busulfan (ilk etkili alkilleyici ajan), hidroksikarbamid ve interferon-alfa (IFN-α) geldi. 1980’lere gelindiğinde, allojenik kök hücre nakli (SCT), uygun hastalar için tercih edilen tedavi haline geldi ve Goldman ve ark. (1982, Lancet) gibi çalışmalar kronik faz KMY’de kullanımını bildirdi. Ancak SCT sınırlıydı, daha küçük aile boyutları nedeniyle gelişmiş ülkelerde %25-30 oranında mevcuttu ve önemli ölüm ve morbidite riskleri taşıyordu.
20. yüzyılın sonları, Talpaz ve ark. (1986, Br. J. Haematol.) tarafından bildirildiği üzere, hastaların %50’sinden azında hematolojik remisyonlar gösteren ve önemli yan etkilere sahip rekombinant IFN-α’yı getirdi. Atılım, 1998’de, Nicholas Lydon, Alex Matter ve Brian Druker tarafından yönetilen ve 1987’de başlayan bir ilaç keşif programı aracılığıyla geliştirilen, BCR-ABL1 proteinini hedef alan ilk tirozin kinaz inhibitörü (TKI) olan imatinib ile geldi. Klinik çalışmalar 1998’de başladı ve imatinib, KMY yönetimini dönüştürerek 2001’de FDA tarafından onaylandı. 2006’ya kadar olan takip verileri, hematoloji kilometre taşlarında belirtildiği gibi imatinib ile tedavi edilen hastalarda %95 beş yıllık sağ kalım gösterdi. Dasatinib ve nilotinib gibi sonraki TKI’ler, imatinib direnci için 2006-2007’de onaylandı ve tedavi seçenekleri genişletildi.
Etki ve Zorluklar
Bu keşifler, KML’yi üç ila beş yıllık tarihsel sağ kalımla ölümcül bir hastalıktan, imatinib tedavisinden iki yıl içinde tam sitogenetik yanıt elde edildiğinde bazı hastaların yaşam sürelerinin genel nüfusa yaklaştığı yönetilebilir kronik bir duruma dönüştürdü. Ancak, Sawyers ve ark. tarafından 2001’de bildirilen, dirence neden olan BCR-ABL mutasyonları gibi zorluklar devam ediyor ve kombinasyon terapileri ve tedavisiz remisyon hedefleri konusunda devam eden araştırmaları gerektiriyor.
İleri OKuma
- Bennett, J.H., (1845). Case of hypertrophy of the spleen and liver in which death took place from suppuration of the blood. Edinburgh Medical and Surgical Journal, 64, 413–423.
- Virchow, R., (1847). Weisses Blut. Archiv für pathologische Anatomie und Physiologie und für klinische Medicin (Virchows Archiv), 1, 563–572.
- Nowell, P.C., & Hungerford, D.A., (1960). A minute chromosome in human chronic granulocytic leukemia. Science, 132(3438), 1497.
- Rowley, J.D., (1973). A new consistent chromosomal abnormality in chronic myelogenous leukemia identified by quinacrine fluorescence and Giemsa staining. Nature, 243(5405), 290–293.
- de Klein, A., van Kessel, A.G., Grosveld, G., Bartram, C.R., Hagemeijer, A., Bootsma, D., Spurr, N.K., Heisterkamp, N., Stephenson, J.R., & Groffen, J., (1982). A cellular oncogene is translocated to the Philadelphia chromosome in chronic myelocytic leukaemia. Nature, 300(5894), 765–767.
- Daley, G.Q., Van Etten, R.A., & Baltimore, D., (1990). Induction of chronic myelogenous leukemia in mice by the P210 bcr/abl gene of the Philadelphia chromosome. Science, 247(4944), 824–830.
- Druker, B.J., Tamura, S., Buchdunger, E., Ohno, S., Segal, G.M., Fanning, S., Zimmermann, J., & Lydon, N.B., (1996). Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nature Medicine, 2(5), 561–566.
- Druker, B.J., Talpaz, M., Resta, D.J., Peng, B., Buchdunger, E., Ford, J.M., Lydon, N.B., Kantarjian, H., Capdeville, R., & Sawyers, C.L., (2001). Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. New England Journal of Medicine, 344(14), 1031–1037.
- Hochhaus, A., & La Rosée, P., (2004). Imatinib therapy in chronic myelogenous leukemia: strategies to avoid and overcome resistance. Leukemia, 18(8), 1321–1331.
- Jabbour, E., & Kantarjian, H., (2012). Chronic myeloid leukemia: 2012 update on diagnosis, monitoring, and management. American Journal of Hematology, 87(11), 1037–1045.










Yorum yazabilmek için oturum açmalısınız.