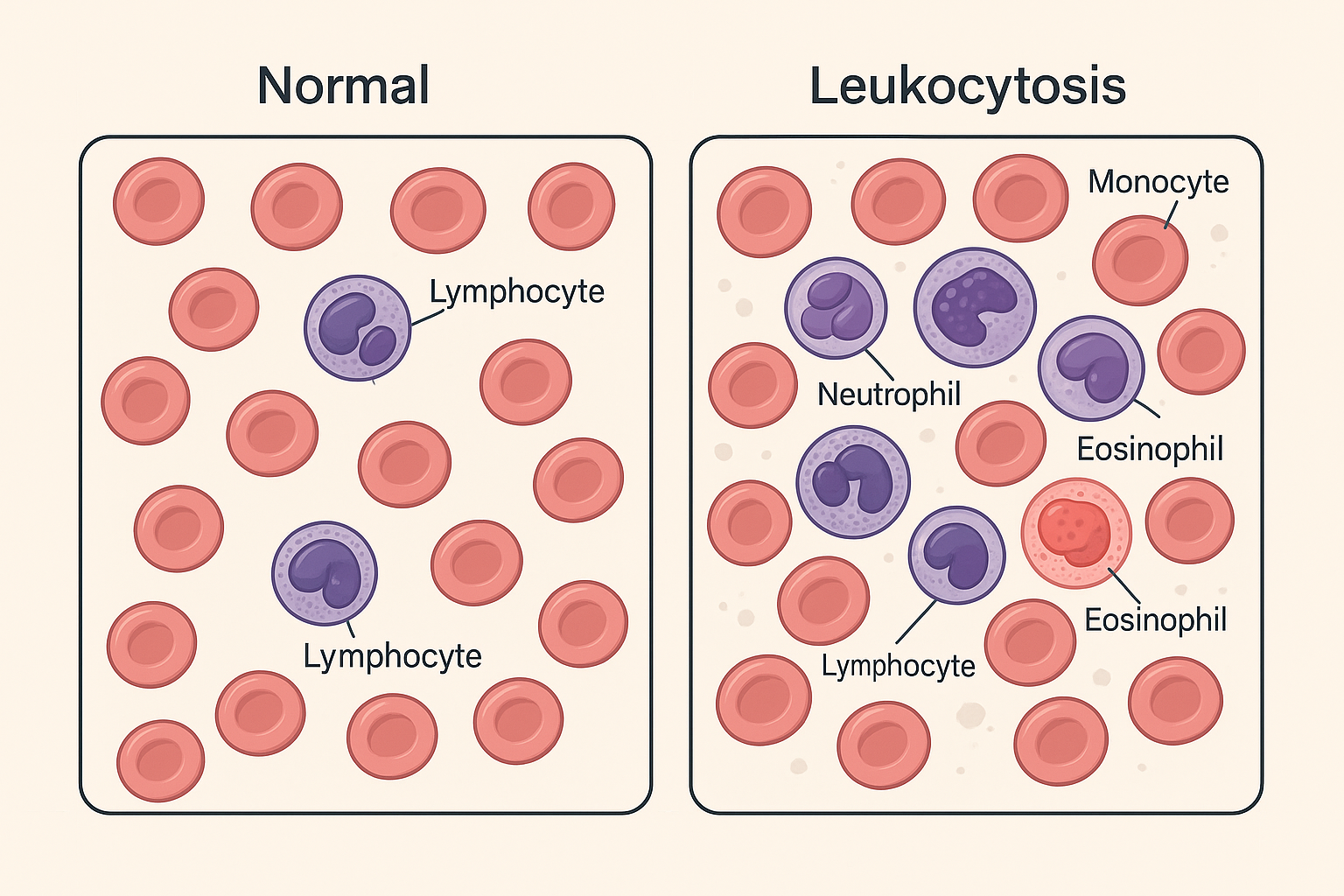
Lökositoz ‒ Kısa Etimoloji
- Leukós (λευκός, Eski Yunanca) → “beyaz”
- Kytos (κύτος, Eski Yunanca) → “hücre, kavite”
- -osis (-ωσις, Eski Yunanca) → “durum, süreç, patolojik artış”
Bu üç kökün Latince birleşimi leucocytosis şeklinde tıbbî terminolojiye girmiş, Türkçede “lökositoz” olarak kullanıma yerleşmiştir; anlamı “beyaz kan hücrelerinde (lökositlerde) artış durumu”dır.
Lökositoz: Tanım ve Genel Kavramlar
- Periferik kandaki toplam lökosit sayısının erişkin kadınlarda > 10 × 10⁹/L, erkeklerde > 11,5 × 10⁹/L olmasına verilen addır.
- Ters durum lökopenidir.
- Çocuklarda fizyolojik üst sınırlar yaşa bağlı olarak belirgin şekilde daha yüksektir.
Sınıflandırma (Diferansiyel Kan Sayımına Göre)
- Nötrofili
- Lenfositoz
- Eozinofili
- Bazofili
- Monositoz

Patofizyoloji ve Özel Terimler
- Sola Kayma (Left Shift): Periferik kanda band formu/öncü granülosit artışı.
- Hiatus Leukaemicus: Kronik miyeloid lösemi (KML) ve bazı akut lösemilerde olgun hücrelerle blaster arasında “ara” serilerin görülmemesi.
- Hiperlökositoz: Lökosit sayısının ≥ 100 × 10⁹/L olması; lökostaz, serebral veya pulmoner mikrosirkülasyon bozuklukları açısından hematolojik acil olarak değerlendirilir.
Etiyoloji – Ayırıcı Tanı
Reaktif (Sekonder) Nedenler
- Akut/kronik bakteriyel, mantar, paraziter enfeksiyonlar (tüberküloz hariç)
- Otoimmün hastalıklar (kollajenozlar, vaskülitler)
- Doku stresi: miyokard enfarktüsü, travma, yanık, şok, akut kanama
- Maligniteler, solid tümörler
- Splenektomi sonrası rebound
- İlaçlar (glukokortikoidler, G-CSF, lityum)
- Gebelik, sigara, yoğun egzersiz, emosyonel stres
Primer (Klonal) Hematolojik Hastalıklar
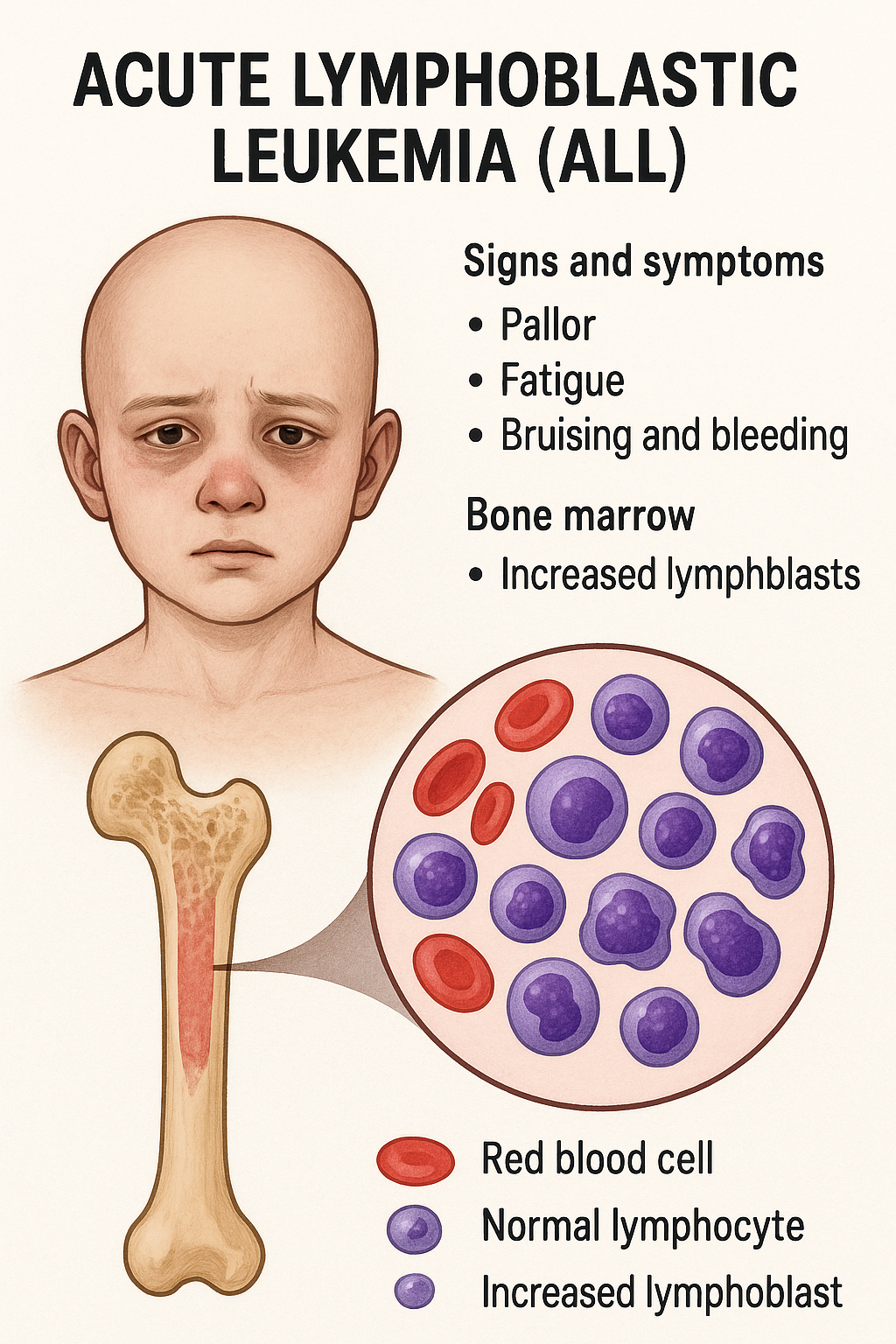
- Akut lösemiler (AML/ALL): Hızlı semptom gelişimi, blastemi; peteşi, anemi, trombopeni eşlik eder.
- Miyeloproliferatif neoplaziler (MPN):
- Kronik miyeloid lösemi (BCR-ABL1 pozitif; imatinib 400 mg/gün ilk basamak)
- Esansiyel trombositemi (ET)
- Polisitemi vera (PV)
- Primer myelofibroz (PMF)
- Kronik idiyopatik nötrofili (CIN): Belirgin tetik olmaksızın persistan nötrofili.
Klinik Bulgular ve Değerlendirme
- Non-spesifik ateş, terleme, kilo kaybı
- Peteşi, ekimoz (eşlik eden trombopeni)
- Anemi semptomları: yorgunluk, dispne
- Tromboemboli veya anormal kanama (özellikle MPN’de)
- Hepatosplenomegali, lenfadenopati
- Lökostaz semptomları: dispne, nörolojik defisit, priapizm
Tanısal Yaklaşım
- Tam kan sayımı + periferik yayma
- Otomatik diferansiyel + mikroskopik doğrulama
- Akış sitometri: blast immunofenotipi
- Sitogenetik/moleküler testler: BCR-ABL1, JAK2 V617F, CALR, MPL, NPM1, FLT3 vb.
- Kemik iliği aspirasyon/biopsi
- Biyokimyasal testler: LDH, ürik asit (hücre dönüşümü göstergesi)

Hiperlökositozun Akut Yönetimi
- Lökoferez ( > 100 × 10⁹/L + semptom )
- Hidroksiüre 50–100 mg/kg/gün hızlı sitoreduksiyon
- Hidratasyon + allopurinol/rasburikaz (tümör lizis profilaksisi)
Miyeloproliferatif Neoplazilerde Tedavi Prensipleri
- Bazal Tedavi: Asetilsalisilik asit 100 mg/gün (trombofilaktik)
- Sitoredüktif Endikasyonlar (HT-54 Kriterleri): Yaş > 60 yıl, önceki tromboz, platelets > 1,5 × 10⁶/µL, JAK2-pozitiflik.
- İlaç Seçenekleri:
- Hidroksiüre (ilk tercih; 500–2000 mg/gün bölünmüş)
- Anagrelid (Xagrid®) – seçici trombosit düşürücü; izole trombositozda
- Peg-interferon-α – genç/hamile olgularda; yorgunluk, grip-benzeri yan etkiler
- Ruxolitinib – JAK1/2 inhibitörü; splenomegali, konstitüsyonel semptomlarda
- İzlem: Tedavinin başlamasından sonra 2-3 haftada ilk etki; ilk 4 haftada doz ayarı. Hematolojik kontroller 3–6 ayda bir, gerektiğinde daha sık.
Akut Lösemilerde Özel Noktalar
- Blaster perifere çıktığında “blaster kanda yeri yoktur” prensibiyle kemoterapiye hızla başlanır.
- Lökosit > 100 × 10⁹/L ise lökoferez ± hidroksiüre.
- AML indüksiyon (7+3) veya ALL protokolleri; transfüzyon ve infeksiyon profilaksisi.
Tedavi İzleminde Dikkat Edilecek Yan Etkiler
- İmatinib: periorbital ödem, kas krampları, hepatotoksisite
- Hidroksiüre: makrositoz, bacak ülserleri
- Anagrelid: taşikardi, baş ağrısı
- Interferon-α: grip-benzeri sendrom, depresyon
- Ruxolitinib: sitopeniler, opportunistik infeksiyon riski
Özet Klinik Yaklaşım Algoritması
- Acil mi? Lökosit ≥ 100 × 10⁹/L + semptom → lökoferez/hidroksiüre.
- Periferik Yayma: Blast var mı?
- Evet → akut lösemi değerlendir, kemik iliği + akış sitometri.
- Hayır → reaktif vs MPN ayrımı (JAK2/BCR-ABL1 vs enfeksiyon, inflamasyon).
- Ek Testler: Moleküler, görüntüleme (splenomegali?), koagülasyon.
- Tedavi ve İzlem: Etiyolojiye yönelik + destek (hidratasyon, allopurinol, ASA).
Keşif
İleri Okuma
- Hoffman, R., et al. (2018). Hematology: Basic Principles and Practice (7th ed.). Elsevier.
- Valent, P., et al. (2019). Contemporary consensus on evaluation and treatment of hyperleukocytosis in acute leukemias. Annals of Hematology, 98(1), 25-44.
- Varki, A., & Tabori, U. (2020). Leukocytosis and leukemoid reactions in infection and inflammation. Seminars in Hematology, 57(3), 171-179.
- Barbui, T., et al. (2021). Clinical practice guidelines for the management of polycythemia vera and essential thrombocythemia. Blood Advances, 5(2), 604-619.
- Cervantes, F., & Tefferi, A. (2021). Primary myelofibrosis: 2021 update on diagnosis, risk-stratification and management. American Journal of Hematology, 96(1), 145-162.
- Arber, D. A., et al. (2022). The 5th edition WHO Classification of Myeloid Neoplasms and Acute Leukemias. Leukemia, 36(5), 1253-1274.
- Cortes, J., & Radich, J. (2024). Chronic myeloid leukemia: 2024 update on frontline therapy. Journal of Clinical Oncology, 42(4), 345-356.
- NCCN (2024). Acute Myeloid Leukemia, Version 3.2024. NCCN Clinical Practice Guidelines in Oncology., 1-152.




/endometrial-cancer-symptoms1-5b2191ec312834003601d53f.png)








Yorum yazabilmek için oturum açmalısınız.