Antik Yunancada, πάθος(páthos, “hastalık”) + -λογία(-logía, “bilimi”) → Fransızca pathologie ‘den türemiştir.
Patoloji, kelimenin tam anlamıyla acı çekme çalışmasına dönüşür; Modern tıbba uygulandığı haliyle, hastalıkların incelenmesidir. Virchow, hastalığın hücresel düzeyde ortaya çıktığını iddia etmekte kesinlikle haklıydı, ancak şimdi hücresel rahatsızlıkların, hücrelerin hayatta kalmasını ve davranışını etkileyen moleküllerdeki (genler, proteinler ve diğerleri) değişikliklerden kaynaklandığını anlıyoruz. Bu nedenle, modern patolojinin temeli, hastalıklara yol açan hücresel ve moleküler anormallikleri anlamaktır. Bu anormallikleri normal hücresel yapı ve işlev bağlamında değerlendirmek yararlıdır.
Halk arasında; kulak çınlaması. (Bkz; Tinnitus) (Bkz; aurium)
Sinonim: Enechema, Susurrus aurium.
Tinnitus aurium, bir ses olayı tarafından tetiklenmeyen ses izlenimlerini tanımlamak için kullanılan bir terimdir. Akustik halüsinasyonlar veya işitme sesleri dahil değildir.
Sınıflandırma
Bulgunun nesneleştirilebilirliğine göre
Temel olarak, ayrım algılanabilirliğe göre yapılır:
Nesnel kulak çınlaması
Öznel kulak çınlaması
Her iki form için de farklı nedenler vardır.
Süreye göre
Farklı tinnitus aurium formları sürelerine göre ayırt edilir:
Akut kulak gürültüsü (ortaya çıktıktan sonra 3 aya kadar)
Kronik kulak gürültüsü (3 aydan itibaren).
Bu sınıflandırma farklı patofizyolojik süreçlere değil klinik deneyime dayanmaktadır. Bununla birlikte, prognoz açısından önemlidir: akut kulak sesleri genellikle kronik seyirli kulak çınlamasından daha sık spontan iyileşme veya iyileşme gösterir. Mevcut bilgi durumuna göre, artık bir ‘subakut kulak gürültüsü’ yoktur, çünkü terapi sadece iki zaman tipine göre yapılır.
Eşlik eden bulgulara göre
İşitme kaybı olan kulak çınlaması
İşitme kaybı olmayan kulak çınlaması
Etiyoloji
Semptomatik (‘otojenik’) kulak çınlamasının nedenleri arasında işitme bozukluğu, gürültü hasarı, Meniere hastalığı ve diğer organik hastalıklar bulunur. İşitme kaybına sıklıkla kulak çınlaması eşlik eder. Servikal omurga veya diş çene bölgesindeki sorunlar somatik kulak çınlamasının nedenlerini tetikleyebilir veya yoğunlaştırabilir. Tıbbi nedenlere ek olarak, bunların yarısı şüpheli gürültü ve stresi psikojenik kulak çınlamasının tetikleyicileri olarak etkiledi.
Belirtiler
Kulak çınlamasının ses izlenimleri bireysel olarak çok çeşitlidir ve diğer şeylerin yanı sıra aşağıdaki karaktere sahip olabilir:
Uğultu
Çınlama
Islık
Tıslama
Cızıltı
Çatlak
Bozukluk sabit bir yoğunluğa veya ritmik veya titreşimli bir karaktere sahip olabilir. Hastalar öznel algılarını her zaman gerçek bir sesle birleştirmeyi başaramazlar.
Beyin sapı tümörlerinin dışlama tanısı (örn. Vestibüler schwannom)
Aşağıdaki sebepler elenmelidir;
Akustik halüsinasyonlar
İşitme sesleri
Tedavi
Kulak çınlaması tedavisi, hastalığın nedenine ve süresine ve şiddetine bağlıdır.
Akut kulak çınlamasında kan dolaşımını (örneğin reolojik tedavi), kortikoid tedavisi ve iyonotropik tedavileri teşvik etmek için önlemler alınır. Kesin bir çalışma olmadığı için her ikisi de etkinlikleri konusunda tartışmalıdır. Bu, gıda takviyeleri, antioksidanlar veya hayali ilaçlar (‘işitme hapı’) için daha fazla derecede geçerlidir. Glutamat antagonistleri için etkinliğin zayıf kanıtı vardır.
Kronik kulak çınlaması danışmanlık, bilişsel davranışçı terapi (CBT), alışılmış terapi, duyarsızlaştırma ve kulak çınlaması yeniden eğitimi ile tedavi edilir. Otojenik kulak çınlaması için ör. İşitme kaybını önlemek için, gürültü koruması için uygun önlemler alınmalıdır (kulak çınlaması profilaksisi).
Alternatif tıpta sıklıkla kullanılan bilimsel olarak değerlendirilmemiş prosedürler, ör. ‘Yumuşak lazer’, ‘vagus sinir stimülasyonu’ vb. – kanıt eksikliği nedeniyle kulak çınlamasının rasyonel tedavisinde yeri yoktur.
Guillain-Barré sendromu, periferik sinir sisteminin akut, genellikle tek atağa sahip, sıklıkla şiddetli seyreden, otoimmün kaynaklı bir poliradikulonöropatisidir. Klinik olarak en temel özellik; birkaç gün ile en fazla dört hafta içinde ilerleyen, çoğunlukla simetrik, arefleksi ile birlikte görülen kas güçsüzlüğüdür. Hastalık tipik olarak alt ekstremitelerde başlayan “yukarı doğru tırmanan” (ascending) flasid paralizi ile seyreder, ağır olgularda solunum kaslarına, kraniyal sinirlere ve otonom sinir sistemine yayılabilir.
Patolojik substrat; omurilik sinir kökleri ve ilgili proksimal periferik sinir segmentlerinde demiyelinizasyon ve/veya aksonal hasar, buna eşlik eden inflamatuar hücre infiltrasyonudur. Bu nedenle GBS, akut inflamatuar demiyelinizan poliradikulonöropati (AIDP) ve aksonal varyantları (AMAN, AMSAN) ile birlikte bir spektrum bozukluğu olarak değerlendirilir.
2. Tarihçe ve etimoloji
Hastalık, klinik açıdan 19. yüzyıl ortalarından itibaren tanımlanmaya başlanmıştır. 1859’da Fransız hekim Jean-Baptiste Octave Landry, bacaklardan başlayıp gövdeye ve nadiren kraniyal kaslara yükselen akut flasid paralizi tablosunu “ascendans paralizi” olarak tarif etmiş, bu tablo uzun süre “Landry paralizisi” adıyla anılmıştır.
1916’da Fransız nörologlar Georges Guillain, Jean-Alexandre Barré ve radyolog André Strohl, Birinci Dünya Savaşı sırasında iki askerde görülen benzer tabloyu ayrıntılı olarak tanımlamış, özellikle beyin-omurilik sıvısında (BOS) hücre sayısında artış olmaksızın protein yüksekliği (albüminositolojik dissosiasyon) bulgusunu sistematik biçimde göstermişlerdir. Bu üçlüden Strohl’un adı daha sonra sendrom adlandırmasında geri plana itilmiş, tablo “Guillain-Barré sendromu” şeklinde yerleşmiştir.
Eponim şu şekilde çözümlenebilir:
“Guillain” – Georges Guillain (1876–1961), periferal nöropatiler ve refleks fizyolojisi üzerine çalışan Fransız nörolog.
“Barré” – Jean-Alexandre Barré (1880–1967), klinik nörolojinin erken dönem kurucu isimlerinden biri.
Fransızca telaffuzda “Guillain-Barré” [giyen-baré] şeklinde okunur; uluslararası literatürde İngilizceleştirilmiş “Gillian-Baré” benzeri telaffuzlar da yaygındır.
Terimsel açıdan GBS, başta “akut idiopatik polinörit”, “akut inflamatuar poliradikulonöropati” gibi farklı adlarla da anılmış; demiyelinizan ve aksonal alt tiplerin tanımlanmasıyla birlikte, günümüzde “post-enfeksiyöz, immün aracılı, akut periferal nöropati” çerçevesine oturtulmuştur.
3. Epidemiyoloji
GBS dünya genelinde tüm yaş gruplarında görülmekle birlikte, nadir bir hastalıktır.
Yıllık insidans: yaklaşık 0,8–1,9/100.000 kişi; medyan insidans 1,3/100.000 olarak bildirilmektedir.
Erkeklerde kadınlara göre belirgin bir fazlalık (yaklaşık 1,5–2:1) söz konusudur.
Her yaşta görülebilmekle birlikte, sıklıkla genç erişkinlik ve ileri yetişkinlik döneminde (yaklaşık 20–30 ve 50–60’lı yaşlarda) iki küçük pik tarif edilmektedir.
Çocukluk çağında insidans daha düşüktür (0,4–1,3/100.000), ancak klinik tablo benzer bir seyir gösterir.
Coğrafi dağılım açısından, AIDP tipik olarak Avrupa ve Kuzey Amerika’da baskınken, aksonal varyantlar (AMAN, AMSAN) özellikle Doğu Asya ve Latin Amerika’da daha yüksek oranda görülmektedir. Bu fark, özellikle Campylobacter jejuni suşlarının bölgesel farklılıkları ve çevresel/konak faktörleri ile ilişkilendirilmektedir.
4. Etiyopatogenez ve immünoloji
4.1. Post-enfeksiyöz otoimmün yanıt ve moleküler taklit
GBS’nin temel mekanizması, çoğu vakada enfeksiyon sonrası gelişen yanlış yönlendirilmiş bir bağışıklık yanıtıdır. Hastaların yaklaşık üçte ikisinde, nörolojik semptomlardan 1–3 hafta önce geçirilmiş üst solunum yolu enfeksiyonu veya gastroenterit öyküsü vardır.
Özellikle Campylobacter jejuni kaynaklı enterit, GBS için en iyi tanımlanmış tetikleyicidir ve olguların yaklaşık üçte birinden sorumlu olduğu kabul edilmektedir. Bunun yanında sitomegalovirüs (CMV), Epstein-Barr virüsü (EBV), Mycoplasma pneumoniae, Zika virüsü, HIV ve bazı durumlarda Haemophilus influenzae, influenza ya da diğer solunum yolu virüsleri ile ilişki gösterilmiştir.
Temel kavram moleküler taklit (molecular mimicry) dir:
Patojenlerin yüzey antijenleri (özellikle Campylobacter lipooligosakkaritleri), periferal sinir gangliosidlerine (GM1, GD1a, GQ1b vb.) yapısal olarak benzer epitoplar içerir.
Bakteriyel antijenlere karşı gelişen antikorlar, bu benzerlik nedeniyle sinir hücre zarındaki gangliosidlere de bağlanarak komplement aktivasyonu, makrofaj infiltrasyonu ve sonuçta demiyelinizasyon veya aksonal hasara yol açar.
4.2. Hedef yapılar ve alt tip ilişkisi
AIDP: Başlıca hedef Schwann hücresi ve myelin kılıftır. CD4+ T lenfositler, makrofajlar ve kompleman aracılı antikor yanıtı, segmental demiyelinizasyon ve iletim bloklarına yol açar.
AMAN ve AMSAN: Aksonal membranlar üzerinde bulunan GM1, GD1a gibi gangliosidlere karşı antikorlar preterminal aksonal bölgede kompleman bağımlı hasar oluşturur; bu tabloda demiyelinizasyon ikincil veya minimaldir.
Miller-Fisher sendromu (MFS): Okülomotor sinirler ve propriyoseptif yolların yüzey gangliosidi GQ1b’ye karşı antikorlar karakteristiktir; klinikte oftalmopleji, ataksi ve arefleksi triadıyla seyreder.
4.3. Aşılar, cerrahi ve diğer tetikleyiciler
Bazı aşılar (özellikle 1976 domuz gribi aşı kampanyası), GBS riskinde hafif artışla ilişkilendirilmiş; daha yeni veriler, modern influenza aşıları için bu riskin son derece düşük, mutlak risk artışının milyon doz başına birkaç ek vaka düzeyinde olduğunu göstermektedir. Enfeksiyonun kendisi çoğunlukla aşı riskine kıyasla çok daha güçlü bir tetikleyici olarak kalmaktadır.(Wikipedia)
Büyük cerrahi girişimler, travma, sistemik inflamatuar hastalıklar, nadiren gebelik ve puerperyum da olguların küçük bir kısmında tetikleyici ya da eşlik eden faktörler olarak bildirilmiştir.(NCBI)
5. Patoloji
Nekropsi ve sinir biyopsisi çalışmalarında:
Spinal sinir kökleri ve proksimal periferik sinir segmentlerinde lenfosit ve makrofaj ağırlıklı interstisyel infiltrasyon,
Schwann hücreleri çevresinde ve Ranvier boğumlarında segmental demiyelinizasyon,
Aksonal varyantlarda Wallerian dejenerasyonu,
Endonöral ve perinöral kapillerlerde geçirgenlik artışı ve ödem
saptanmıştır. Demiyelinizan tipte iletim blokları ve remiyelinizasyon odakları, aksonal tipte ise iletim amplitüdlerinde belirgin azalma ile uyumlu histopatolojik bulgular hâkimdir.
6. Klinik özellikler ve seyrin evreleri
6.1. Başlangıç ve prodromal dönem
Hastaların önemli bir kısmı, nörolojik belirtilerden birkaç gün önce:
Halsizlik, miyalji, subfebril ateş,
Hafif paresteziler (özellikle ayak parmaklarında ve ellerde),
Bel veya bacakta derin, sızı tarzında ağrı
tarif eder. Bu dönemde klinik muayene çoğu zaman belirgin bulgu vermeyebilir.
6.2. İlerleme (progresyon) fazı
Kardinal klinik özellikler:
İlerleyici, simetrik kas güçsüzlüğü:
Tipik olarak alt ekstremiteden başlar, saatler–günler içinde proksimale ve üst ekstremitelere, gövde ve yüz kaslarına yayılır.
Flasid, areflektik paralizi söz konusudur.
Derin tendon reflekslerinin kaybı (arefleksi): Tanı için anahtar bulgulardan biridir; bazı erken vakalarda hiporefleksi görülebilir.
Duyusal belirtiler: Hafif distal paresteziler, vibrasyon ve pozisyon duyusunda bozulma sık; ağır objektif duyu kaybı genellikle demiyelinizan tipte belirgindir.
Otonom disfonksiyon: Taşikardi, hipertansif veya hipotansif ataklar, ortostatik hipotansiyon, kardiyak aritmiler, terleme bozuklukları, mesane-barsak işlevlerinde değişiklik görülebilir.
Ağrı: Özellikle bel ve bacaklarda, gece artan radiküler ağrı çok yaygındır.
Semptomların başlamasından sonra güçsüzlük genellikle 2–4 hafta içinde en ağır düzeyine (nadir) ulaşır; tanım gereği GBS’de progresyon süresi 4 haftayı aşmaz. Daha uzun süren progresyon, kronik inflamatuar demiyelinizan polinöropati (CIDP) lehinedir.
6.3. Platon ve iyileşme evresi
Nadirden sonra 1–2 hafta süren stabil dönem (plato) izlenir.
Ardından, aylar hatta yıllar sürebilen yavaş bir iyileşme süreci başlar; remiyelinizasyon ve aksonal rejenerasyon bu dönemde klinik düzelmenin temelini oluşturur.
Tam nörolojik iyileşme, olguların yaklaşık %60–80’inde; hafif–orta düzeyde kalıcı sekeller %10–20’sinde; ağır kalıcı özürlülük veya respiratuvar destek gereksinimi ise daha küçük bir grupta izlenir.
Avrupa ve Kuzey Amerika’da GBS vakalarının çoğunu oluşturur.
Klinik tablo klasik şekildedir: simetrik, distaldan proksimale yayılan güçsüzlük, arefleksi, duyusal belirtiler ve sıklıkla otonom bulgular.
Elektrofizyolojide iletim hızlarında yavaşlama, distal latanslarda uzama, iletim blokları, temporal dispersiyon ve F-dalga latanslarında belirgin artış saptanır.
7.2. Akut motor aksonal nöropati (AMAN)
Özellikle Çin, Japonya, Meksika gibi bölgelerde sık; sıkı Campylobacter jejuni ilişkisi vardır.
Klinik: Saf motor tutulum; duyusal belirtiler minimal veya yoktur.
Elektrofizyolojide motor aksiyon potansiyeli amplitüdlerinde azalma, ancak belirgin demiyelinizasyon bulgusu olmaması tipiktir.
7.3. Akut motor-duyusal aksonal nöropati (AMSAN)
Hem motor hem duyusal aksonların ağır tutulduğu, hızlı ilerleyen ve sıklıkla daha kötü prognozlu bir formdur.
Özellikle yoğun bakım ihtiyacı ve uzun rehabilitasyon süresi ile ilişkilidir.
7.4. Miller-Fisher Sendromu (MFS)
Klinik triad: oftalmopleji + ataksi + arefleksi, çoğu vakada ekstremite güçsüzlüğü minimal veya yoktur.
GQ1b antikorları yüksek titrede pozitifliği ile karakterizedir.
Zaman zaman klasik GBS ile örtüşen olgular oluşturur (MFS-GBS overlap).
7.5. Diğer varyantlar
Faringeal-servikal-brakiyal zayıflık formu
Saf kraniyal polinöritis
Bickerstaff beyin sapı ensefaliti (MFS spektrumu içinde kabul edilir)
Bu varyantlarda klinik topografya farklı olsa da altta yatan immünolojik mekanizmalar çoğu kez klasik GBS ile süreklilik gösterir.
8. Tanı
GBS tanısı, başta klinik olmak üzere elektrofizyolojik ve BOS bulgularının bütüncül değerlendirilmesine dayanır. Güncel EAN/PNS kriterleri, progresif güçsüzlük, arefleksi ve elektrofizyolojik demiyelinizasyon bulgularının kombinasyonunu temel alır.
8.1. Klinik temel kriterler
İki veya daha fazla ekstremitede ilerleyici motor güçsüzlük
Derin tendon reflekslerinde belirgin azalma ya da yokluk
Semptom başlangıcından nadire kadar sürenin 4 haftadan kısa olması
Başlangıçta belirgin ateş olmaması (enfeksiyon döneminden sonra nörolojik belirtilerin başlaması tipiktir)
8.2. Destekleyici klinik bulgular
Simetrik tutulum eğilimi
Belirgin sfinkter disfonksiyonunun olmaması (olması durumunda ayırıcı tanı genişletilir)
BOS’ta albüminositolojik dissosiasyon
Elektrofizyolojide demiyelinizan veya aksonal polinöropati bulguları
İlerleyen günlerde reflekslerin kaybının daha da belirginleşmesi
8.3. BOS incelemesi
Tipik bulgu albüminositolojik dissosiasyondur:
Protein artışı (>0,55 g/L ya da laboratuvar referans değerinin üzerinde)
Normal ya da hafif artmış lökosit sayısı (<10/mm³; daha yüksek hücre sayıları alternatif tanıları düşündürür).
Protein artışı semptom başlangıcından sonraki ilk birkaç günde hafif olabilir; çoğu olguda ikinci haftadan itibaren belirginleşir.
8.4. Elektrofizyolojik çalışmalar
Sinir iletim çalışmaları (NCS) ve iğne elektromiyografisi, alt tip ayrımı ve tanı doğrulaması için kritik önemdedir:
Aksonal tipte motor ve/veya duyusal aksiyon potansiyeli amplitüdlerinde belirgin düşme;
Bu bulgular, EAN/PNS kriterlerine göre “kesin”, “olası” GBS ayrımını da belirler.
8.5. Ayırıcı tanı
GBS’nin ayırıcı tanısında:
Spinal kord lezyonları (transvers miyelit, kompresyon)
Akut motor nöron hastalıkları
Myastenia gravis krizi ve botulizm
Kene paralizisi
Porfiri, toksik nöropatiler, difteri nöropatisi
Akut inflamatuar myopatiler
gibi çok sayıda tablo yer alır; bu nedenle dikkatli nörolojik muayene, görüntüleme ve laboratuvar değerlendirmesi gerekir.
9. Tedavi ve klinik yönetim
GBS tedavisinde iki ana eksen vardır: (1) yaşamı tehdit eden komplikasyonların önlenmesi için yoğun destekleyici bakım, (2) immünomodülatör spesifik tedavi (IVIG veya plazmaferez). Güncel kılavuzlar, bu iki immün tedavinin etkinlik açısından birbirine yakın olduğunu, ancak beraber kullanılmaması gerektiğini vurgular.
9.1. Destekleyici bakım
Solunum izlemi ve desteği:
Vital kapasite, FVC, negatif inspiratuvar basınç gibi parametreler yakından izlenir.
Hastaların %20–30’unda mekanik ventilasyon gerekebilir; özellikle hızlı ilerleme, bulbar tutulum ve ciddi otonom disfonksiyon varlığında risk yüksektir.
Otonomik instabilite yönetimi:
Aritmi ve kan basıncı dalgalanmaları için sürekli kardiyak monitörizasyon,
Gereğinde kısa etkili antihipertansifler veya vazopressörler.
Tromboembolizm ve bası yarası profilaksisi:
Düşük molekül ağırlıklı heparin, elastik çoraplar, erken pasif hareket,
Düzenli pozisyon değişimi ve bası alanlarının korunması.
Beslenme ve yutma:
Bulbar tutulum ve aspirasyon riski olan hastalarda nazogastrik ya da erken perkütan gastrostomi ile enteral beslenme.
Ağrı yönetimi:
Nöropatik ağrı için gabapentinoidler, trisiklik antidepresanlar veya karbamazepin;
Akut kas ağrıları için nonsteroid antiinflamatuvar ilaçlar veya gerekirse opioidler.
9.2. İmmünomodülatör tedavi
9.2.1. İntravenöz immünoglobulin (IVIG)
Doz: 0,4 g/kg/gün, ardışık 5 gün (toplam 2 g/kg).
Etkinliği, plazmaferezle benzer düzeydedir; uygulanması daha kolay ve birçok merkezde tercih edilen ilk tedavidir.
Semptom başlangıcından itibaren ilk 2 hafta içerisinde başlanması önerilir; en büyük fayda genellikle ilk 7 gün içinde tedavi edilenlerde görülür.
Yan etkiler: Baş ağrısı, aseptik menenjit, tromboemboli riski, nadiren böbrek fonksiyon bozukluğu.
Güncel uzman görüşleri, rutin olarak ikinci IVIG kürünün uygulanmasını önermemekte; kötü prognozlu olgularda bile ikinci kürün yarardan çok yan etki riskini artırdığına dair veriler bildirilmektedir.
9.2.2. Plazmaferez (terapötik plazma değişimi)
Tipik rejim: 1–1,5 plazma volümünü içeren 4–5 değişim, 7–14 gün içine yayılmış şekilde; daha hafif olgularda 2 değişimlik kısa protokoller de kullanılabilir.
Mekanizma: Otoantikorlar, kompleman bileşenleri ve immün komplekslerin dolaşımdan uzaklaştırılması.
İmmobil ve hemodinamik olarak dalgalı hastalarda santral venöz kateter gereksinimi, hipotansiyon, enfeksiyon vb. komplikasyon riskleri nedeniyle dikkatli hasta seçimi gerekir.
9.2.3. Kortikosteroidler ve diğer ajanlar
Oral veya intravenöz kortikosteroid monoterapisi, büyük randomize çalışmalarda GBS’de anlamlı fayda göstermediği için önerilmez.
IVIG + plazmaferez kombinasyonu da rutin olarak önerilmez; ardışık uygulamanın net bir ek faydası gösterilmemiş, maliyet ve yan etki yükünü artırdığı bildirilmiştir.
Deneysel düzeyde monoklonal antikorlar, kompleman inhibitörleri ve diğer immünomodülatörler üzerine çalışmalar sürmektedir; henüz standart pratiğin parçası değillerdir.
9.3. Rehabilitasyon
Erken dönemde pasif eklem hareketleri, postüral destek ve basit oturma-ayakta durma egzersizleri, kontraktür ve kas atrofisini önlemeye yardımcı olur.
İyileşme fazında, düşük yoğunluklu, yorucu olmayan kas güçlendirme ve yürüme eğitimi, sinir rejenerasyonuna eşlik eden plastik uyumu destekler; aşırı egzersizle kas yorgunluğu ve “overwork weakness” riskine dikkat edilmelidir.
10. Prognoz ve uzun dönem sonuçlar
GBS mortalitesi modern yoğun bakım olanaklarıyla yaklaşık %3–7 arasındadır; ölümlerin başlıca nedenleri ventilatör ilişkili komplikasyonlar, ağır otonom disfonksiyona bağlı kardiyak olaylar ve sepsisdir.
Hastaların çoğu ilk yıl içinde bağımsız yürüyüş yetisine geri döner; ancak:
%15–20 oranında kalıcı hafif–orta güçsüzlük, ayak düşmesi veya ince motor becerilerde zayıflık,
%20–40 oranında kronik yorgunluk ve efor intoleransı,
Aksonal formlar (AMAN/AMSAN), ileri yaş, çok hızlı progresyon, erken ventilasyon gereksinimi ve ağır otonom disfonksiyon, kötü prognoz belirteçleri olarak öne çıkar.
11. Kronik inflamatuar demiyelinizan polinöropati (CIDP) ile ilişki
CIDP, klinik ve patolojik açıdan GBS ile birçok ortak özellik paylaşan ancak kronik seyirli bir polinöropatidir. Progresyon süresi 8 haftayı aşar veya relapslarla dalgalanır; tedavide IVIG ve plazmaferez yanında steroid ve uzun dönem immünsupresif ajanlara da yer verilir. Bu iki tablonun, akut ve kronik uçları aynı immünolojik spektrum içinde oluşturduğu düşünülmektedir.
12. Evrimsel tıp perspektifi
GBS’yi evrimsel açıdan değerlendirmek, bağışıklık sisteminin ince ayarlı dengesinin, modern çevresel koşullar altında nasıl kırılganlaştığını anlamak için öğretici bir örnek sunar:
Yüksek risk–yüksek fayda dengesi: Bağışıklık sistemi, patojenleri hızlı ve etkili tanıyıp yok etmek için geniş bir antijen tanıma repertuvarı ve güçlü efektör mekanizmalar geliştirmiştir. Ancak bu geniş repertuvar, bazı mikrobiyal antijenlerin ev sahibi dokulardaki moleküllerle benzeşmesine (moleküler taklit) izin verir; GBS, bu dengenin patojen lehine değil, otoimmünite lehine kaydığı durumların tipik örneğidir.
Patojen evrimi ve konak yanıtı: Campylobacter jejuni gibi bakteriler, yüzey yapılarında evrimsel baskı altında çeşitlenirken, bazı suşlar gangliosid benzeri yapılar kazanmıştır. Bu değişiklikler, bakteri için bağışıklıktan kaçma veya konak hücrelere tutunma avantajı sağlayabilirken, insanda GBS riskini artıran moleküler taklit zeminini oluşturur.
Çevresel değişim ve enfeksiyon spektrumu: Global seyahat, gıda üretim zincirlerindeki değişiklikler ve iklimle ilişkili vektör kaymaları (örn. Zika virüsü salgınları), belirli bölgelerde GBS vakalarında kümelenmeler ve yeni tetikleyicilerin ortaya çıkmasına yol açmıştır. Bu da bağışıklık sistemimizin evrimsel olarak daha az maruz kaldığı patojenlerle etkileşimini artırarak nadir otoimmün komplikasyonların görünürlüğünü güçlendirmiştir.
Aşılama ve risk algısı: Evrimsel perspektif, bireysel nadir yan etkilere odaklanmanın, popülasyon düzeyinde enfeksiyon yükü ve buna bağlı GBS dahil ağır nörolojik komplikasyonların önlenmesiyle kazanç dengesini çoğu zaman yanlış algılamaya yol açabileceğini hatırlatır. Modern aşıların GBS için mutlak risk artışı çok düşükken, hedefledikleri enfeksiyonlar GBS için güçlü tetikleyiciler olmaya devam etmektedir.
Keşif
Guillain-Barré sendromu (GBS) tarihsel olarak, önce klinik sahnede “garip ama tanımlanabilir” bir tablo olarak belirmiş, ardından adım adım immünolojinin, nöropatolojinin ve elektrofizyolojinin merkezine doğru çekilmiş bir hastalık öyküsüdür. Bugün GBS’yi; kollarda, bacaklarda ve kimi zaman gövde ve kraniyal kaslarda hızla gelişen güçsüzlüğe ve duyusal bozukluğa yol açan, kendi kendini sınırlayan akut bir polinöropati olarak tanımlıyoruz. Bu akut polinöropati, çoğu kez solunum yolu ya da gastrointestinal sistem enfeksiyonlarının ardından ortaya çıkan, bağışıklık aracılı bir süreç olarak kabul edilmektedir.
Bu öykünün klasik başlangıç noktası 1916 yılıdır. Birinci Dünya Savaşı’nın kaotik ortamında, üç Fransız hekim – nörolog Georges Guillain, nörolog Jean Alexandre Barré ve radyolog/elektrofizyolog André Strohl – cepheden gelen iki askerde son derece benzer bir tablo saptar. Her iki askerde de akut motor güçsüzlük, arefleksi (derin tendon reflekslerinin kaybı) ve buna eşlik eden ama ağır olmayan duyusal yakınmalar vardır. Guillain, Barré ve Strohl’u asıl etkileyen bulgu ise lomber ponksiyonla aldıkları beyin omurilik sıvısında (BOS) ortaya çıkar: protein düzeyi belirgin olarak artmıştır, ancak hücre sayısı normaldir. Yani BOS’ta o güne kadar pek tanımlanmamış bir kombinasyon gözlenir: “yüksek protein + normal hücre sayısı.” Bugün “albüminositolojik dissosiasyon” olarak adlandırdığımız bu özellik, GBS için adeta imzasal bir biyokimyasal işaret hâline gelir.
Bu iki asker vakasının ayrıntılı klinik betimlemesi ve BOS bulgularının vurgulanmasıyla birlikte, sendrom 1916’da literatüre girer. Başlangıçta bu tablo, esas olarak periferik sinir sisteminin demiyelinizan bir bozukluğu olarak görülür ve “akut inflamatuar demiyelinizan polinöropati” (AIDP) terimi giderek yerleşir. Yani uzun bir süre boyunca GBS denince, akla omurilik sinir kökleri ve periferik sinir liflerinin miyelin kılıfında gelişen inflamatuar hasar gelir; aksonların kendisi çoğunlukla ikincil ya da daha az belirgin kabul edilir.
Ancak 1980’lerden itibaren bu çerçeve sarsılmaya başlar. Özellikle Doğu Asya ve Latin Amerika’dan bildirilen olgularda, klinik olarak GBS’ye benzeyen fakat elektrofizyolojik olarak belirgin demiyelinizasyon göstermeyen, buna karşın motor aksiyon potansiyeli amplitüdlerinde ağır azalma saptanan vakalar tanımlanır. Bu olgularda patolojik inceleme, asıl hasarın miyelinden çok aksonlarda olduğunu gösterir. Böylece GBS’nin yalnızca demiyelinizan bir hastalık olmadığı, “akut aksonal tip” bir formun da bulunduğu anlaşılır ve bu form giderek “akut motor aksonal nöropati (AMAN)” adıyla anılmaya başlar. Kısa süre sonra motor ve duyusal aksonları birlikte tutan akut motor ve duyusal aksonal nöropati (AMSAN) formu tanımlanır; GBS’nin tekil bir hastalıktan çok, ortak bir immünolojik tema etrafında toplanan heterojen bir sendromlar ailesi olduğu fikri güçlenir.
Bu tarihsel genişlemenin bir diğer önemli adımı, klinik açıdan “farklı ama akraba” bir tablonun tanımlanmasıdır. Oftalmopleji (göz kaslarında felç), ataksi (denge ve koordinasyon bozukluğu) ve arefleksi triadı ile seyreden, ekstremite güçsüzlüğünün bazen çok hafif ya da hiç olmadığı bir sendrom, Miller Fisher sendromu (MFS) adıyla literatüre girer. Zaman içinde MFS’nin de GBS ile patofizyolojik bir spektrum oluşturduğu, hatta GBS’nin “kraniyal ağırlıklı” bir varyantı olarak kabul edilebileceği anlaşılır. Buna ek olarak faringeal-servikal-brakiyal varyant (PCB) gibi, farinks, boyun ve omuz kuşağını baskın olarak tutan alt tipler de tanımlanır. Böylece tarihsel olarak tek bir tanımla başlayan sendrom, klinik, elektrofizyolojik ve anatomik yayılım kalıplarına göre dallanan zengin bir GBS alt tipleri haritasına dönüşür.
Patogenez cephesinde ise gelişme, nöroloji tarihinin daha yeni dönemine, immünolojik tekniklerin ve moleküler biyolojinin olgunlaştığı yıllara denk gelir. GBS’nin, yalnızca “enfeksiyonu takip eden bir sinir iltihabı” olmadığının; aslında sinir membranlarındaki belirli glikolipidlere karşı gelişen otoantikorların merkezde olduğu bir bağışıklık bozukluğu olduğunun ortaya konması, dönüm noktalarından biridir. Özellikle aksonal tip GBS ve MFS olgularında, sinir liflerinin membranında bulunan GM1, GD1a, GQ1b gibi gangliosidlere yönelen antikorların sık görüldüğü gösterilir. Bu antikorlar iki temel yolla hasar yaratabilir: Bir yandan kompleman sistemini aktive ederek sinir lifinde doğrudan yapısal hasara neden olurlar; diğer yandan, nodus Ranvier düzeyinde sodyum kanalları ve iletim aparatına bağlanarak sinir iletimini fonksiyonel olarak bloke edebilirler. Böylece, 1916’da yalnızca “yüksek BOS proteini ve arefleksi ile giden akut felç” olarak tarif edilen sendrom, yüzyılın sonuna gelindiğinde moleküler taklit, anti-gangliosid antikorları ve kompleman aracılı sinir hasarı üçgeninde açıklanan bir otoimmün nöropati modeline dönüşmüş olur.
Tarih içinde tedavi yaklaşımı da aynı oranda evrilmiştir. İlk on yıllarda tedavi daha çok destekleyici bakım ile sınırlıyken, 20. yüzyılın son çeyreğinde plazma değişimi (plazmaferez) ve intravenöz immünoglobulin (IVIG), hastalığın seyrini gerçek anlamda değiştiren iki temel müdahale olarak sahneye çıkar. Plazmaferez, dolaşımdan otoantikorları ve immün kompleksleri uzaklaştırarak; IVIG ise çoklu mekanizmalar (otoantikorların nötralizasyonu, reseptör blokajı, kompleman modülasyonu vb.) üzerinden bağışıklık yanıtını yeniden ayarlayarak hastalığın şiddetini ve süresini azaltabilir. Buna rağmen, bazı GBS vakaları refrakter seyreder veya ağır otonomik tutulum, yaygın aksonal hasar ve yaşlılık gibi faktörler nedeniyle kötü prognoz gösterir; bu durum günümüzde hâlâ yeni tedavi stratejilerine duyulan ihtiyacı canlı tutmaktadır.
Bugün GBS, klinik özellikleri, elektrofizyolojik bulguları ve immünolojik profilleri farklılaşan alt tipleriyle heterojen bir hastalık olarak kabul edilir. En sık görülen form hâlâ miyelin kılıfı hedef alan akut inflamatuar demiyelinizan polinöropati (AIDP)’dir; bunun yanında AMAN, AMSAN, MFS ve PCB varyantları, sendromun ne kadar geniş bir spektrum oluşturduğunu gösterir. Çoğu olgunun öncesinde Campylobacter jejuni, sitomegalovirüs, Epstein-Barr virüsü, Zika virüsü gibi bir enfeksiyon öyküsünün bulunması, tarihsel olarak Guillain, Barré ve Strohl’un yalnızca gözlemledikleri, ama adını koyamadıkları bağı “enfeksiyon → çapraz reaksiyon → otoimmünite” zinciriyle somutlaştırır.
Tanı artık; klinik kriterler, sinir iletim çalışmaları ve BOS analizini bir araya getiren sistematik algoritmalara dayanmakta; tedavi ise destekleyici bakımın yanı sıra IVIG veya plazma değişimi ile standartlaştırılmış protokoller üzerinden yürütülmektedir. Yine de, tüm bu tarihsel ilerlemeye karşın, GBS’ye bağlı mortalite – başlıca solunum yetmezliği ve hareketsizliğe bağlı komplikasyonlar nedeniyle – hâlâ yaklaşık %5 civarındadır ve azımsanmayacak sayıda hasta kalıcı güçsüzlük, ağrı ya da sakatlıkla yaşamına devam etmektedir.
Böyle bakıldığında GBS’nin tarihi, 1916’da üç Fransız hekimin iki askerde fark ettiği akut felç tablosundan, günümüzde alt tipleri, antikor profilleri ve hedef molekülleri ayrıntılı biçimde çözümlenen, fakat hâlâ yeni tedaviler aranan karmaşık bir otoimmün nöropati evrenine uzanan kesintisiz bir hikâye olarak okunabilir.
İleri Okuma
Landry O (1859) Note sur la paralysie ascendante aiguë. Gazette Hebdomadaire de Médecine et de Chirurgie 6:472–474.
Guillain G, Barré JA, Strohl A (1916) Sur un syndrome de radiculonévrite hyperalbuminose du liquide céphalo-rachidien sans réaction cellulaire. Bull et Mem Soc Med Hop Paris 40:1462–1470.
Haymaker W, Kernohan JW (1949) The Landry-Guillain-Barré syndrome: a clinicopathologic report of fifty fatal cases. Arch Neurol Psychiatry 61:577–618.
Miller Fisher C (1956) Ophthalmoplegia, ataxia and areflexia: a syndrome. N Engl J Med 255:57–65. doi:10.1056/NEJM195607122550201
Griffin JW, Li CY, Macko C, et al. (1996) Guillain-Barré syndrome: pathogenesis of axonal injury. Ann Neurol 39(1):17–28. doi:10.1002/ana.410390107
Rees JH, Soudain SE, Gregson NA, Hughes RAC (1995) Campylobacter jejuni infection and Guillain-Barré syndrome. N Engl J Med 333:1374–1379. doi:10.1056/NEJM199511233332102
Hadden RDM, Cornblath DR, Hughes RAC, et al. (1998) Electrodiagnostic classification of Guillain-Barré syndrome. Ann Neurol 44(5):780–788. doi:10.1002/ana.410440512
Yuki N (1999) Pathogenesis of axonal Guillain-Barré syndrome: antiganglioside antibodies and molecular mimicry. J Infect Dis 179(Suppl 2):S150–S153. doi:10.1086/513850
Jacobs BC, Rothbarth PH, van der Meché FGA, et al. (1998) The spectrum of antecedent infections in Guillain-Barré syndrome: a case-control study. Neurology 51(4):1110–1115. doi:10.1212/WNL.51.4.1110
Hughes RAC, Wijdicks EFM, Barohn R, et al. (2003) Practice parameter: immunotherapy for Guillain-Barré syndrome: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 61(6):736–740. doi:10.1212/01.WNL.0000085054.43699.7E
Kuwabara S (2004) Guillain-Barré syndrome: epidemiology, pathophysiology and management. Drugs 64(6):597-610.
Hughes RAC, Cornblath DR (2005) Guillain-Barré syndrome. Lancet 366(9497):1653-1666.
van Doorn PA, Ruts L, Jacobs BC (2008) Clinical features, pathogenesis, and treatment of Guillain-Barré syndrome. Lancet Neurol 7(10):939–950. doi:10.1016/S1474-4422(08)70215-1
Dimachkie MM, Barohn RJ (2013) Guillain-Barré syndrome and variants. Neurol Clin 31(2):491-510.
Willison HJ, Jacobs BC, van Doorn PA (2016) Guillain-Barré syndrome. Lancet 388(10045):717–727. doi:10.1016/S0140-6736(16)00339-1
Doets AY, Verboon C, van den Berg B, et al. (2018) Regional variation of Guillain-Barré syndrome. Brain 141(10):2866–2877. doi:10.1093/brain/awy227
Shahrizaila N, Lehmann HC, Kuwabara S (2021) Guillain-Barré syndrome. Lancet 397(10280):1214–1228. doi:10.1016/S0140-6736(21)00517-1
Willison HJ, Yuki N (2022) Peripheral neuropathies and antiganglioside antibodies. Neurotherapeutics 19:562–577. doi:10.1007/s13311-022-01231-3
van den Berg B, Walgaard C, Drenthen J, et al. (2024) Advances in diagnosis and treatment of Guillain-Barré syndrome. Nat Rev Neurol 20(1):25–42. doi:10.1038/s41582-023-00826-5
Kusunoki S (2016) History of Guillain-Barré syndrome. Clin Exp Neuroimmunol 7(4):318-329.
Shahrizaila N, Lehmann HC, Kuwabara S (2021) Guillain-Barré syndrome. Lancet 397(10280):1214-1228.
Laman JD, Füger P (2022) Molecular mimicry in Guillain-Barré syndrome: expanding the concept. Trends Immunol 43(9):734-747.
Finsterer J, Scorza FA (2022) Infectious triggers and immune dynamics in Guillain-Barré syndrome. Int J Mol Sci 23(22):14222.
Nguyen TP, Taylor RS (2023) Guillain-Barré Syndrome. StatPearls. StatPearls Publishing, Treasure Island (FL).
van Doorn PA et al. (2023) European Academy of Neurology/Peripheral Nerve Society guideline on diagnosis and treatment of Guillain-Barré syndrome. J Peripher Nerv Syst 28(3):175-228.
Wiegers EJA, van den Berg B (2025) New insights in the immune treatment of Guillain–Barré syndrome. Curr Opin Neurol 38(5):xxx-xxx.
Serebral korteksin (beyin kabuğunun) altında subkortikal patolojik hasara yol açan vasküler değişikliklerin (arterioskleroz) neden olduğu bir beyin hastalığıdır.
Beyaz beyin maddesine verilen hasarın neden olduğu küçük damar vasküler demansının bir şeklidir.
İlk olarak Jena’daki İsviçreli nörolog Otto Ludwig Binswanger (1852-1929) tarafından tanımlandı. 1894’te Otto Binswanger tarafından tanımlandı ve Alois Alzheimer ilk kez 1902’de ‘Binswanger hastalığı’ ifadesini kullandı. Bununla birlikte, Olszewski, 1962’de başlayan bu hastalığın günümüzdeki araştırmasının çoğuyla karşılanmaktadır.
“Hepatik Ensefalopati” (HE) terimi, çeşitli kökenlerden kaynaklanan karaciğer hastalığına bağlı olarak ortaya çıkan nörolojik ve psikopatolojik semptomların bir koleksiyonunu ifade eder (Bakınız; Hepatik, ensefal-o-pati). Karaciğer zararlı maddelerin vücuttan atılmasında önemli bir rol oynar. Karaciğer hasar gördüğünde bu maddeler birikebilir ve beyin fonksiyonlarını etkileyerek HE semptomlarına yol açabilir.
Epidemiyoloji
Sınıflandırma
Hepatik Ensefalopatinin üç ana türü vardır:
Tip A: Bu, karaciğerin detoksifikasyon fonksiyonunu kaybettiği akut karaciğer yetmezliği nedeniyle ortaya çıkar. Bu, beyin ortamını destekleyen ve koruyan astrositlerde sıvı birikmesi nedeniyle beynin şiştiği bir durum olan beyin ödemine yol açabilir.
Tip B: Bu, karaciğerin vücudun dolaşım sistemi tarafından bypass edilmesi olan portosistemik şant nedeniyle oluşur. Bu herhangi bir parankimal karaciğer hastalığı olmadan gerçekleşebilir.
Tip C: Bu, kronik karaciğer hastalığı, portal hipertansiyon ve portosistemik şant ile birlikte karaciğer sirozunda ortaya çıkar.
HE ayrıca alt türlere ayrılabilir:
Epizodik HE
Kalıcı HE
Minimal HE (MHE)
Tip B ve C HE’nin patofizyolojisi, karaciğerin detoksifikasyon fonksiyonunun bozulmasını içerir ve bu da amonyak gibi nörotropik maddelerin portal ven halkasından kan dolaşımına taşmasına yol açar. Bu maddeler daha sonra beyin fonksiyonlarını etkileyerek HE semptomlarına yol açabilir. Kendiliğinden veya ameliyat sonucu portosistemik şantın oluşması da portal hipertansiyona yol açabilir ve karaciğer fonksiyonunun daha da bozulmasına neden olabilir. HE gelişimine katkıda bulunabilecek diğer hızlandırıcı faktörler arasında enfeksiyon (örn. spontan bakteriyel peritonit), gastrointestinal kanama, ilaçlar, azotemi, elektrolit dengesizliği, kabızlık ve diyetteki protein yükü yer alır.
HE tedavisi durumun ciddiyetine ve şekline bağlıdır. Laktuloz, amino asitler ve rifaximin gibi ilaçların kullanımını içerebilir veya ciddi vakalarda karaciğer nakli gerekebilir.
This content is available to members only. Please login or register to view this area.
Teşhis
Hepatik ensefalopati (HE), beyni etkileyen ve karaciğer hastalığıyla ilişkili karmaşık bir durumdur. HE tanısı, nörolojik bozuklukların diğer nedenlerini dışlamayı ve ensefalopatinin ciddiyetini değerlendirmeyi amaçlayan bir dizi test ve değerlendirmeyi içerir. Kullanılan teşhis araç ve yöntemlerinden bazıları şunlardır:
Koma hastalarında apopleksiyi dışlamak için kafatası BT.
Hipoglisemiyi dışlamak için kan şekeri seviyelerinin belirlenmesi.
Hiperamonyemiyi tespit etmek için plazma amonyak seviyelerinin ölçümü.
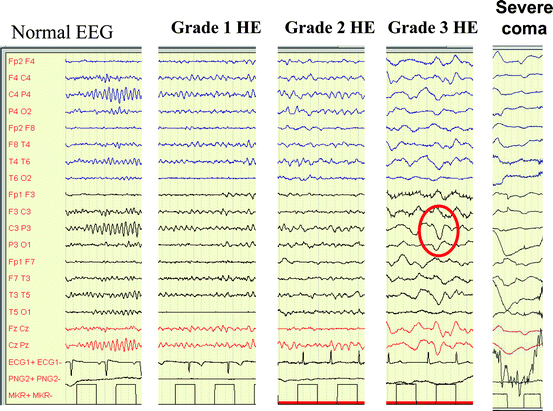
Kramp potansiyellerini belirlemek için EEG.
Ensefalopatiyi evrelemek ve minimal formları tanımak için EASL veya AASLD gibi psikometrik test prosedürleri. HE’nin ciddiyetini daha iyi nesnelleştirmek ve sınıflandırmak için West Haven sınıflandırma sistemi kullanılır. Bu sistem, durumu klinik semptomlara göre aşağıdaki şekilde aşamalandırır:
Aşama I: Uyuşukluk, konsantrasyon güçlüğü, ruh hali değişimleri, basit aritmetik problemlerini çözmede problemler ve titreme başlangıcı da dahil olmak üzere ince motor becerilerde bozulma ile karakterizedir.
Aşama II: Artan uyku hali, apati, dizartri, sınırlı zamansal yönelim ve EEG değişikliklerinin başlangıcı ile işaretlenmiştir.
Aşama III: Bu aşamadaki hastalar çoğunlukla uyur ancak uyandırılabilirler. Uyandıktan sonra tutarsız konuşma, artan kas gerginliği (spastisite) ve yeni başlayan fetor hepaticus sergileyebilirler.
Aşama IV: Bu, hastaların kornea ve kas reflekslerinin söndüğü ve ağrılı uyaranlara yanıt vermediği karaciğer komasında olduğu en şiddetli aşamadır.
Şiddet
Semptom
Asteriisks
EEG (/sn.)
Ammoyiak arteriyel (μg/dl)
I
Huzursuzluk, unutkanlık, titreme, öfori veya kaygı, toplama hesaplamasında başarısız olma
nadir
7–8
151–200
II
Uyuşukluk, yönelim bozukluğu, hafif kişilik değişikliği, kötüleşen çıkarma hesaplamaları
düzenleşen
5–7
201–250
III
Somnolans, sopor, belirgin oryantasyon bozukluğu
sık
3–5
251–300
IV
Koma, piramit işareti
düzenli
< 3
> 300
West Haven sınıflamasına ek olarak minimal hepatik ensefalopati (MHE) de tanımlanabilmektedir. MHE, klinik-nörolojik muayene sırasında semptom göstermeyen ancak nöropsikometrik testlerle tespit edilebilen bilişsel bozuklukların olduğu HE olarak tanımlanır. MHE’nin tedavi gerektiren bir durum olup olmadığı konusunda devam eden tartışmalar vardır.
West Haven sınıflandırmasının MHE ve evre I’i topluca “klinik altı HE” veya “gizli HE” olarak anılırken II ila IV. evreler “açık HE” olarak adlandırılır.
Tedavi
Hepatik ensefalopati, akut karaciğer yetmezliği veya kronik karaciğer hastalığının bir sonucu olarak ortaya çıkabilen karmaşık bir durumdur. Tedavi yaklaşımı, durumun altında yatan nedene bağlı olarak değişir.
Akut karaciğer yetmezliği vakalarında çeşitli tedavi seçenekleri mevcuttur:
Karaciğer Nakli: Karaciğer hasarı ciddi ve geri döndürülemez ise karaciğer nakli gerekli olabilir.
Mannitol ile Osmoterapi: Mannitol, serebral ödemin azaltılmasına yardımcı olmak için bir ozmotik diüretik olarak kullanılabilir.
Kontrollü Hipotermi: Bu, beyin hasarı riskini azaltmaya yardımcı olmak için vücut sıcaklığının düşürülmesini içerir.
Kronik karaciğer hastalığı olan hastalar için tedavi yaklaşımı, altta yatan durumun yönetilmesine ve komplikasyonların önlenmesine odaklanır:
Tetikleyici Faktörlerin Belirlenmesi ve Ortadan Kaldırılması: Bu, enfeksiyonlar veya ilaçlar gibi hepatik ensefalopatiye katkıda bulunabilecek faktörlerin ele alınmasını içerir.
İlave Katabolik Fonksiyonlardan Kaçınma: Hastalara, durumu daha da kötüleştirebileceği için gıda alımını kısıtlamamaları tavsiye edilir.
Azotlu Maddelerin Bağırsaktan Ortadan Kaldırılması: Bu, kandaki amonyak seviyelerinin azaltılmasına yardımcı olan laktulozun oral veya klinik uygulamasıyla sağlanabilir.
Ornitin Aspartat ile Hiperammoneminin Azaltılması: Bu amino asit takviyesi kandaki amonyak seviyelerinin azaltılmasına yardımcı olur.
Dallanmış Zincirli Amino Asitlerin Oral Uygulaması: Bu amino asitler karaciğer fonksiyonu için önemlidir ve hepatik ensefalopati semptomlarının azaltılmasına yardımcı olabilir.
Bağırsakta Amonyak Oluşturan Bakterilerin Azaltılması: Bu, bağırsakta amonyak üreten bakteri seviyelerini azaltmaya yardımcı olan bir antibiyotik olan rifaximin alınarak sağlanabilir.
Genel olarak, hepatik ensefalopatinin tedavisi, durumun altında yatan nedeni ele alan ve hastanın yaşam kalitesini iyileştirmek için semptomları yöneten kapsamlı bir yaklaşım gerektirir.
Tarihçe
Hepatik ensefalopati (HE), ciddi karaciğer hastalığı olan hastalarda ortaya çıkan nöropsikiyatrik bir sendromdur. Hasarlı karaciğer tarafından temizlenemeyen toksinlerin kanda birikmesinden kaynaklanır. Bu toksinler kan-beyin bariyerini geçerek beyni etkileyerek kafa karışıklığı, yönelim bozukluğu, uyuşukluk ve koma gibi çeşitli semptomlara yol açar.
HE’nin bilinen ilk tanımı M.Ö. 5. yüzyılda Hipokrat’a kadar uzanır. Hipokrat, sarılığı (cildin ve gözlerin sararması) olan kişilerin aynı zamanda kafa karışıklığı, deliryum ve koma yaşadığı bir durumu tanımladı.
17. yüzyılda İtalyan doktor Giovanni Battista Morgagni HE’nin karaciğer hastalığından kaynaklandığını öne süren ilk kişiydi. Karaciğer hastalığı olan birçok kişinin aynı zamanda nörolojik semptomlar da yaşadığını gözlemledi.
19. yüzyılda Alman doktor Nikolai Vladimirovich Eck, köpeklerde portosistemik şant oluşturarak HE’nin ilk deneysel modelini oluşturdu. Portosistemik şant, portal ven ile sistemik dolaşım arasında karaciğeri bypass eden bir bağlantıdır. Eck’in deneyleri, portosistemik şantların kanda toksin birikmesine ve nörolojik semptomların gelişmesine yol açabileceğini gösterdi.
20. yüzyılda HE’nin anlaşılması ve tedavisinde birçok ilerleme kaydedildi. 1950’lerde HE’yi tedavi etmek için ilk ilaçlar geliştirildi. Bu ilaçlar HE’de rol oynadığı düşünülen zehirli bir madde olan amonyak üretimini azaltmak için tasarlandı.
1960’lı yıllarda ilk karaciğer nakli yapıldı. Karaciğer nakli HE’nin en etkili tedavisidir ancak her hasta için tam şifa değildir.
yüzyılda Alman şair Heinrich Heine HE hastası olduğu biliniyordu. Heine 59 yaşında HE’den öldü.
yüzyılda Amerikalı yazar ve mizahçı Dorothy Parker HE hastası olduğu biliniyordu. Parker 73 yaşında HE nedeniyle öldü. Komik Gerçekler
HE’ye bazen “çırpma titreme sendromu” denir çünkü HE’li bazı kişilerin ellerinde kanat çırpma titremesi gelişir.
HE’ye bazen “asteroid ensefalopatisi” de denir çünkü HE’li bazı kişiler asteroit halüsinasyonları geliştirir.
HE’li bazı kişiler dondurma yiyerek semptomlarını azaltabileceklerini bulmuşlardır. Dondurmanın beyindeki sinirleri uyuşturmaya ve iltihabı azaltmaya yardımcı olduğu düşünülüyor.
Diğerleri ise müzik dinleyerek semptomlarını azaltabileceklerini bulmuşlardır. Müziğin beyni rahatlatmaya ve stresi azaltmaya yardımcı olduğu düşünülmektedir.
Vilstrup, H., Amodio, P., Bajaj, J., et al. (2014). Hepatic encephalopathy in chronic liver disease: 2014 Practice Guideline by the European Association for the Study of the Liver and the American Association for the Study of Liver Diseases. Journal of Hepatology, 61(3), 642-659. doi: 10.1016/j.jhep.2014.05.042
Ferenci, P., Lockwood, A., Mullen, K., Tarter, R., Weissenborn, K., & Blei, A. T. (2002). Hepatic encephalopathy—definition, nomenclature, diagnosis, and quantification: final report of the working party at the 11th World Congresses of Gastroenterology, Vienna, 1998. Hepatology, 35(3), 716-721. doi: 10.1053/jhep.2002.31250
Bajaj, J. S., Etemadian, A., Hafeezullah, M., et al. (2007). Testing for minimal hepatic encephalopathy in the United States: An AASLD survey. Hepatology, 45(3), 833-834. doi: 10.1002/hep.21573
Randolph, C., Hilsabeck, R., & Kato, A. (2009). Neuropsychological and neurophysiological assessments of hepatic encephalopathy. Journal of Hepatology, 50(5), 1069-1075. doi: 10.1016/j.jhep.2009.02.018
Weissenborn, K. (1998). Diagnosis of minimal hepatic encephalopathy. Journal of Hepatology, 28(1), 129-134. doi: 10.1016/S0168-8278(98)80280-1
Bajaj, J. S., Cordoba, J., Mullen, K. D., Amodio, P., Shawcross, D. L., Butterworth, R. F., & Morgan, M. Y. (2011). Review article: the design of clinical trials in hepatic encephalopathy – an International Society for Hepatic Encephalopathy and Nitrogen Metabolism (ISHEN) consensus statement. Alimentary pharmacology & therapeutics, 33(7), 739-747. Link to the reference
Prasad, S., Dhiman, R. K., & Duseja, A. (2010). Lactulose and hepatic encephalopathy: systematic review and meta-analysis of randomized controlled trials. Journal of gastroenterology and hepatology, 25(6), 1029-1036. Link to the reference
Sharma, P., & Sharma, B. C. (2011). Rifaximin and hepatic encephalopathy: an important message for clinicians. Journal of clinical gastroenterology, 45(6), 554. Link to the reference
Hastalık Tipi: Akut sistemik vaskülit; bilinmeyen etiyolojiye sahip, enfeksiyöz özellikler gösteren, ancak esasen immün kaynaklı olduğu düşünülen bir hastalıktır.
Yaş Grubu: Genellikle 5 yaş altındaki bebeklerde ve yürümeye başlayan çocuklarda görülür.
Kardiyak Komplikasyon Riski: Özellikle koroner arterlerde anevrizma gelişimi gibi ciddi kalp komplikasyonlarıyla ilişkilidir.
Epidemiyoloji
Görülme Sıklığı: 5 yaş altı Kafkas çocuklarında yıllık insidans yaklaşık 9 / 100.000’dir.
Coğrafi Dağılım: Sanayileşmiş ülkelerde daha yaygın olarak bildirilmiştir.
Etiyoloji
Kesin Nedeni Bilinmemektedir.
Hipotezler:
İmmünolojik: Vasküler endotel hücrelerine karşı otoimmün yanıt.
İnfeksiyöz: Viral ya da bakteriyel süperantijenler aracılığıyla tetiklenen bağışıklık yanıtı.
Çevresel: Troposferik rüzgarlarla taşınan Candida sp. kaynaklı mikotoksinler ile ilişkili olabileceği düşünülmektedir.
Patogenez ve Morfoloji
Vasküler Tutulum: Küçük ve orta kalibre arterlerde nekrotizan vaskülit.
En çok etkilenen damarlar: Koroner arterler.
Histopatolojik Özellikler: İnflamatuvar hücre infiltrasyonu, damar duvarında nekroz, fibrinoid değişiklikler.
Belirti ve Semptomlar
Majör Bulgular (≥5 gün süren ateş + en az 4 majör kriter):
Konjonktivit: Bilateral, seröz, pürülan olmayan.
Oral ve Mukozal Değişiklikler: Çilek dili, çatlamış dudaklar, hiperemik farinks.
Ekstremite Değişiklikleri:
Akut dönemde palmar ve plantar eritem, şişlik.
Subakut dönemde parmaklarda deskuamasyon.
Döküntü (Ekzantem): Polimorfik; makülopapüler, ürtiker benzeri, gövde ve ekstremitelerde.
Lenfadenopati: Özellikle servikal, >1,5 cm çaplı, genellikle tek taraflı.
Ek Semptomlar:
Genel durum bozukluğu
Enantem (ağız içi lezyonlar)
El ve ayaklarda kızarıklık ve ödem
İshal, kusma, karın ağrısı gibi gastrointestinal belirtiler
Artralji ve artrit
Tanı
Klinik Tanı Kriterleri:
Ateş ≥ 5 gün ve yukarıdaki 5 kriterden en az 4’ü.
Kriterlerin tümü mevcutsa, tanı için ateş süresi şartı aranmayabilir.
Eksik Kawasaki Sendromu: Klinik bulgular tam değilse, koroner arter anevrizması varlığı tanıya götürebilir.
Laboratuvar Bulguları:
Lökositoz: Sola kaymalı nötrofili
Trombositoz: 2. haftadan itibaren, 3. haftada pik
Anemi: Normokrom normositik
CRP ve AÇH: Belirgin artış
Karaciğer enzimleri: Transaminaz yüksekliği
İdrar: Steril piyüri (%33)
Kardiyak Değerlendirme:
Ekokardiyografi: Koroner arter anevrizmaları, kalp kapak patolojileri
Ateş sonrası 4–6 hafta: 3–5 mg/kg/gün (antitrombotik doz)
Dirençli Vakalar:
doz IVIG
Kortikosteroidler
İnfliksimab gibi anti-TNF ajanları (dirençli olgularda)
Takip ve İzlem
Ekokardiyografi: Başlangıçta, 2. haftada, 6. haftada ve uzun vadeli izlemde.
EKG ve Kardiyak Markerler: Miyokardit ve infarktüs riski açısından.
Trombosit sayımı ve inflamasyon markerları: Klinik iyileşmenin takibi için.
Prognoz
İyileşme: Uygun ve erken tedavi ile çoğu olgu tamamen iyileşir.
Uzun Dönem İzlem: Koroner arterlerde kalıcı değişiklik riski nedeniyle gereklidir.
Risk Faktörleri:
Tanıda gecikme
Ateş başlangıcından sonra geç IVIG uygulaması
Dirençli vakalarda yetersiz yanıt
Koroner arter tutulumunun varlığı
Keşif
1. İlk Gözlem ve Klinik Tanım (1961)
Kawasaki hastalığı, ilk olarak Ocak 1961 tarihinde Japon pediatrist Tomisaku Kawasaki (川崎 富作) tarafından Tokyo’daki Japon Kızılhaç Tıp Merkezi’nde, döküntü, yüksek ateş, konjonktivit ve oral mukozal değişiklikler gösteren dört yaşındaki bir erkek çocukta gözlemlendi.
Kawasaki, bu olguyu ilk başta klasik enfeksiyon hastalıkları sınıfına sokamadı. Çünkü hastalık kızıl, kızamık, Stevens-Johnson sendromu gibi bilinen döküntülü hastalıklardan farklı seyrediyordu.
Bu vakadan sonra, Tomisaku Kawasaki 6 yıl boyunca benzer belirtilere sahip 50 çocuk vakası daha tanımlayarak 1967 yılında Japonca olarak ilk bilimsel yayınını yaptı: Kawasaki T. (1967). Acute febrile mucocutaneous lymph node syndrome with specific desquamation of the fingers and toes in children. Arerugi, 16(3), 178–222.
2. Kardiyak Tutulumun Fark Edilmesi (1967–1970)
Kawasaki ve meslektaşları 1967 sonrası 23 vakayı detaylı şekilde inceleyerek elektrokardiyografi kullanımıyla hastalıkta kardiyak tutulum olabileceğini raporladılar.
Bu 23 hastadan 11’inde (%48) elektrokardiyografik anomaliler saptandı ve bunun üzerine hastalığın sistemik vaskülit komponenti olduğu anlaşılmaya başlandı.
Bu dönemde Kawasaki, hastalığı “Mucocutaneous Lymph Node Syndrome (MCLS)” olarak adlandırdı ve etiyolojinin bilinmediğini ancak immünolojik bir zemin olabileceğini savundu.
3. Uluslararası Tanınma ve İngilizce Literatüre Giriş (1974)
Kawasaki hastalığı, Japonya dışındaki bilim çevrelerinin dikkatini ilk kez 1974 yılında çekti. Bu yılda hastalığın İngilizce literatürdeki ilk tanımı yayımlandı ve böylece uluslararası pediatri camiası hastalık hakkında bilgi sahibi oldu: Landing, B. H., Larson, E. J. (1974). Are some infantile polyarteritis cases examples of a new disease entity?Pediatrics, 54(5), 624–628.
4. Batı Dünyasında İlk Gözlem ve Tanısal Paralellik (1976)
1976 yılında, Amerikalı araştırmacı William Melish ve arkadaşları, Hawaii’de yaşayan Japon kökenli 16 çocukta Kawasaki hastalığına birebir benzeyen bir sendromu tanımladılar.
Melish ve Kawasaki, birbirlerinden bağımsız olarak günümüzde halen geçerli olan tanı kriterlerini geliştirdiler.
Bu durum, hastalığın yalnızca Japonya’ya özgü bir durum olmadığını, küresel bir klinik varlık olduğunu kanıtladı: Melish, M. E., Hicks, R. M., Larson, E. J. (1976). Infantile polyarteritis nodosa: A unique disease with coronary artery involvement. The Journal of Pediatrics, 88(2), 240–245.
5. Geriye Dönük Patolojik İncelemeler ve Tarihöncesi Bulgular
1980’li yıllarda yapılan retrospektif patolojik analizlerde, 1870 yılında ölen 7 yaşında bir çocuğa ait korunmuş kalp dokusu, koroner arterlerde üç anevrizma ve trombotik lezyonlar içeriyordu.
Bu bulgular, Kawasaki hastalığına özgü patomorfolojik değişikliklerle örtüştüğü için hastalığın muhtemelen 20. yüzyıl öncesinde de mevcut olduğunu ortaya koydu.
6. Tanı Kargaşası: Poliarteritis Nodoza ile Karışma
Batı tıbbında 20. yüzyılın başlarına kadar bildirilen bazı infantil poliarteritis nodoza vakalarının, gerçekte Kawasaki hastalığı olduğu düşünülmektedir.
Özellikle ölümcül seyreden ve koroner anevrizma gösteren vakaların, Kawasaki sendromunun tanımlanmasından önce uygun tanı araçlarının olmaması nedeniyle başka vaskülitlerle karıştırıldığı anlaşılmıştır.
7. Modern Dönem ve Epidemiyolojik Yayılım
1980’lerden itibaren, hastalık Japonya, Kore, Tayvan gibi Doğu Asya ülkelerinde yüksek prevalans göstermeye başladı.
Amerika Birleşik Devletleri ve Avrupa ülkelerinde de sıklıkla teşhis edilmeye başlandı.
Günümüzde Kawasaki hastalığı, gelişmiş ülkelerde çocuklarda edinilmiş kalp hastalığının en yaygın nedeni olarak kabul edilmektedir ve bu bağlamda akut romatizmal ateşin yerini almıştır: McCrindle, B. W., et al. (2017). Diagnosis, treatment, and long-term management of Kawasaki disease: A scientific statement for health professionals from the American Heart Association. Circulation, 135(17), e927–e999.
İleri Okuma
Kawasaki, T. (1967). Acute febrile mucocutaneous lymph node syndrome with specific desquamation of the fingers and toes in children. Arerugi, 16(3), 178–222.
Landing, B. H., Larson, E. J. (1974). Are some infantile polyarteritis cases examples of a new disease entity?Pediatrics, 54(5), 624–628.
Melish, M. E., Hicks, R. M., Larson, E. J. (1976). Infantile polyarteritis nodosa: A unique disease with coronary artery involvement. The Journal of Pediatrics, 88(2), 240–245.
Shulman, S. T., Rowley, A. H. (1995). Does Kawasaki disease have a retroactive history?Pediatric Research, 37(1), 1–6
Rowley, A. H., & Shulman, S. T. (1998). Pathogenesis and management of Kawasaki disease. Expert Reviews in Molecular Medicine, 1998, 1–13.
Newburger, J. W., et al. (2004). Diagnosis, Treatment, and Long-Term Management of Kawasaki Disease: A Statement for Health Professionals. Circulation, 110(17), 2747–2771.
Burns, J. C., & Glodé, M. P. (2004). Kawasaki syndrome. The Lancet, 364(9433), 533–544.
Uehara, R., & Belay, E. D. (2012). Epidemiology of Kawasaki disease in Asia, Europe, and the United States. Journal of Epidemiology, 22(2), 79–85.
Rodó, X., et al. (2014). Tropospheric winds from northeastern China carry the etiologic agent of Kawasaki disease from its source to Japan. Proceedings of the National Academy of Sciences, 111(22), 7952–7957.
McCrindle, B. W., et al. (2017). Diagnosis, treatment, and long-term management of Kawasaki disease: A scientific statement for health professionals from the American Heart Association. Circulation, 135(17), e927–e999.
Doğuştan hipotiroidizm, hipotiroidizmin özel bir biçimidir, embriyonik gelişimdeki bir bozukluktan kaynaklanır ve inaktif tiroid bezinin yetersizliği ile sonuçlanır.(Bkz;Konjenital)
Epidemiyoloji
1: 3,200 doğum (kistik fibrozise benzer)
Bu nedenle en sık görülen konjenital endokrinopatidir.
Patofizyoloji
Konjenital hipotiroidizm çoğu durumda organ gelişimindeki bir bozukluktan kaynaklanır. Az gelişmiş tiroid bezi, çok az veya hiç tiroid hormonu üretilmediği anlamına gelir. Sebepleri bugün hala bilinmemektedir (2020). Farklı genlerdeki mutasyonların olası tetikleyiciler olduğu söylenir.
Athroidi, tiroid displazisi veya ektopya
Daha az yaygın: hormon biyosentezinde veya insizyonda kusur;
Çok nadir: tiroid hormon direnci (T3 reseptör defekti).
Klinik
Konjenital hipotiroidizmi olan yenidoğanlarda klinik tablo çok belirsiz ve en geniş anlamda kas hipotansiyonu ve olası uzun süreli bir ikterus ile sınırlıdır.
Geç etkileri
Kalkan bezinin yetersiz işlevi erken bebeklik döneminde tedavi edilmezse, aşağıdaki komplikasyonlar ortaya çıkar:
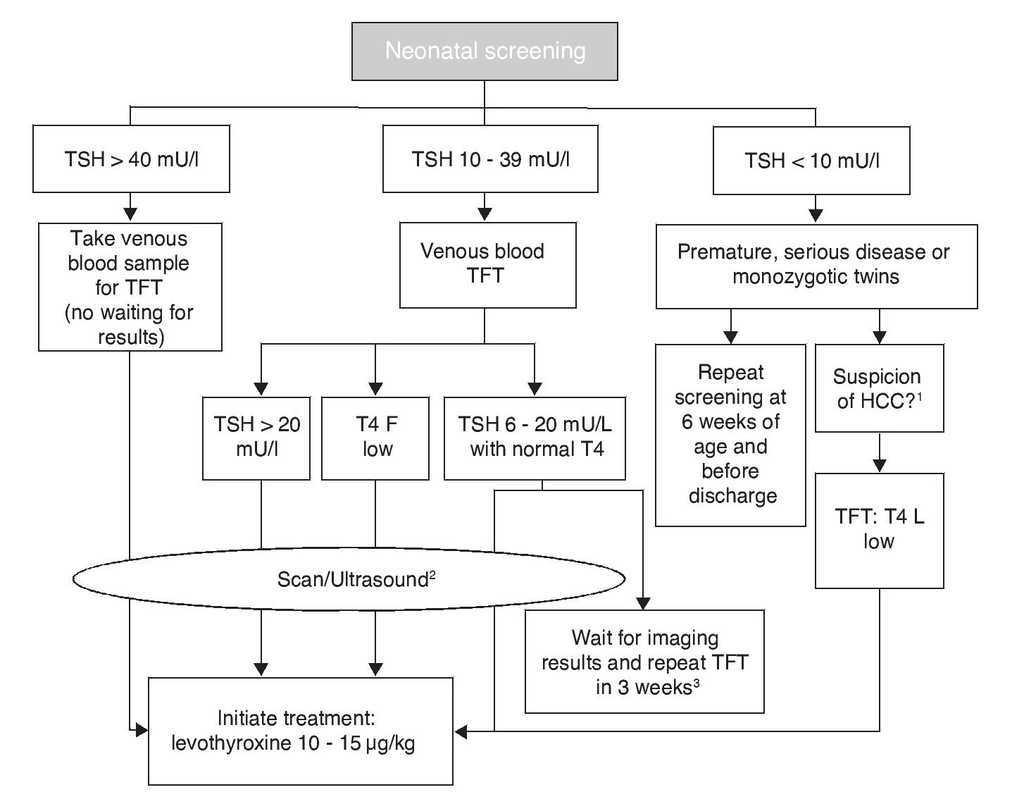
Yenidoğan taraması sayesinde, yaşamın ilk günlerinde konjenital hipotiroidizm tespit edilir. Kılcal topuk kanı alarak ve tarama kartına uygulayarak, metabolizma laboratuvarı tirotropin değerini (TSH ‘u önemli ölçüde artırır) ölçerek az aktif tiroid hakkında sonuçlar çıkarabilir. Test sonucu pozitifse, sıvı kandaki tiroid hormonlarının kantitatif bir tayini izler.
Tedavi
Oral L-tiroksin ikamesi ile tedavi, tanıdan hemen sonra başlatılmalıdır, çünkü ikincil hastalıkları önlemenin tek yolu budur.
Sinonim: Motor neuron diseases, motor neurone diseases (MNDs)
Motor nöronları etkileyen, hiyerarşik tiplerine ve ulaştığı yere göre klinik belirtilerin ve tanıların belirlenmesinde homojen olmayan hastalık grubudur.
Motor nöron hastalıklarında sınırlanma net değildir. Örn: ICD-11 kaydedilen hastalıklardan motor nöron hastalıkları; Kalıtsal Spastik Parapleji (HSP).
ICD-11’e göre, Motor nöron hastalıklarına aşağıdaki hastalıklar dahildir:
Amyotrofik lateral skleroz (ALS)
Progresif ampul felci (PBP)
Progresif psödo-polar felç
Primer lateral skleroz (PLS)
Juvenil primer lateral skleroz (JPLS)
Ilerleyici kas atrofisi (PMA)
Monomelik amiyotrofi (MMA)
Motoneuron Hastalığı Madras (MMND)
Bu hastalıklar üst motor nöronları, alt motor nöronları veya her iki hücre grubunu etkileyebilir.
Epidemiyoloji
Motor nöron hastalıkları hem çocuklarda hem de yetişkinlerde görülür. Çocukları etkileyenler kalıtsal veya ailesel olma eğilimindedir; semptomları doğumda veya yürümeyi öğrenmeden önce ortaya çıkar.
Yetişkinleri etkileyenler 40 yaşından sonra ortaya çıkma eğilimindedir
Klinik
Belirti ve semptomlar spesifik hastalığa bağlıdır, ancak motor nöron hastalıkları tipik olarak hareketle ilişkili bir grup semptom olarak kendini gösterir.
Yavaş yavaş gelişirler ve üç aydan fazla sürüp, kötüleşirler. Çeşitli kas güçsüzlükleri görülür, kas krampları ve spazmları oluşabilir.
Merdiven çıkma (efor) ile nefes almakta, uzanırken (ortopne) nefes almakta güçlük çekebilir, hatta kasları solumakta solunum yetmezliği yaşayabilir.
Konuşma güçlüğü (dizartri), yutma güçlüğü (disfaji) ve aşırı tükürük üretimi (siyalor) gibi bulbar semptomları da ortaya çıkabilir.
Duyum veya hissetme yeteneği tipik olarak etkilenmez. Duygusal bozukluk (ör. Psödobulbar etkisi), bilişsel ve davranışsal değişiklikler (örneğin, akıcılık, karar verme ve hafızada problemler) görülür.
Farklı motor nöron hastalıklarında çeşitli kas güçsüzlükleri görülür. Zayıflık simetrik veya asimetrik olabilir ve distal, proksimal veya her ikisi de olan vücut kısımlarında ortaya çıkabilir. Statland ve arkadaşlarına göre, motor nöron hastalıklarında görülen üç ana zayıflık modeli vardır:
Duyusal kayıp olmadan simetrik zayıflık (örneğin PMA, PLS)
Simetrik fokal orta hat proksimal zayıflığı (boyun, gövde, bulbar tutulumu; örn. ALS, PBP, PLS)
Motor nöron hastalıkları, üst ve alt motor nöron tutulumu açısından bir spektrumdadır. Bazılarında sadece alt veya üst motor nöron bulguları bulunurken, diğerleri her ikisinin bir karışımına sahiptir. Alt motor nöron (LMN) bulguları kas atrofisi ve fasikülasyonları içerir ve üst motor nöron (UMN) bulguları hiperrefleksi, spastisite, kas spazmı ve anormal refleksleri içerir.
Saf üst motor nöron hastalıkları veya sadece UMN bulguları olanlar PLS’yi içerir.
Saf alt motor nöron hastalıkları veya sadece LMN bulguları olanlar PMA’yı içerir.
Hem UMN hem de LMN bulgularına sahip motor nöron hastalıkları arasında ailesel ve sporadik ALS bulunur.
Teşhis
Düşük motor nöron bulguları (örneğin kas kaybı, kas seğirmesi), üst motor nöron bulguları (örn. Tempolu refleksler, Babinski refleksi, Hoffman’ın refleksi, artmış kas tonusu) veya her ikisi kombinasyonlu şekilde olabilir.
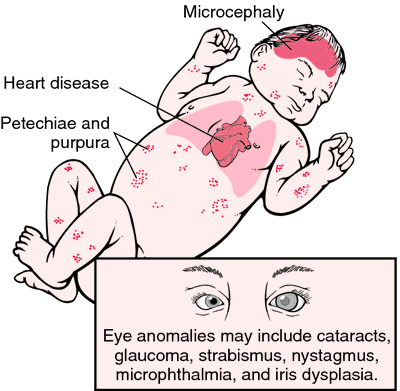
Anne kızamıkçık ile enfekte olduğunda intrauterin kızamıkçık enfeksiyonuna bağlı konjenital hastalık. Maternal enfeksiyon zamanına bağlı olarak, malformasyonlar özellikle gözler, kalp, SSS ve kulakta, aynı zamanda lokal bozukluklarda ortaya çıkar. Kalıcı bir tedavi mümkün değildir, ancak aktif bağışıklama yoluyla etkili profilaksi yoluyla önüne geçilebilir.
Kaynak: https://img2.tfd.com/mk/R/X2604-R-26.png
Epidemiyoloji
Kızamıkçık embriyopatisi 10.000 doğumda 1 insidansa sahiptir. Bu düşük insidans, kapsamlı aşılamadan (MMR aşısı) ve tutarlı prenatal tanıdan kaynaklanmaktadır.
Fizyopatoloji
Bir kadın hamilelik sırasında kızamıkçık virüsü ile enfekte olursa, vücuttaki genel yayılımının bir parçası olarak plasenta yoluyla fetüse yayılabilir. Kızamıkçık virüsü ile enfeksiyon, fetüsteki hücre bölünmesini ve farklılaşma süreçlerini bozar ve yenidoğanda kürtaj veya embriyopati ve kızamıkçık sendromuna yol açar. Kızamıkçık genellikle hafifse, hamile kadın asemptomatik olabilir.
Hamilelik sırasında daha erken kızamıkçık enfeksiyonu ortaya çıkarsa, kızamıkçık embriyopatisi daha olasıdır. Embriyopati riski bu nedenle ilk üç aylık dönemde en yüksektir. Anneye gebeliğin 12. haftasından önce enfekte olursa, vakaların yaklaşık üçte birinde kızamıkçık embriyopatisi meydana gelir. Hamileliğin ilk ayında, kızamıkçık virüsünün% 60’ı fetüse bulaşır. Risk, üçüncü aydan sonra sürekli olarak düşer, dördüncü ayda% 10’luk bir artık riske düşmektedir.
Gebeliğin ilerleyen bölümlerinde bulaşma riski nispeten düşüktür. Bununla birlikte, son üç aylık dönemde meydana gelen enfeksiyonlar da embriyoya zarar verebilir.
Klinik
Kızamıkçık embriyopatisi çeşitli malformasyonlara neden olabilir, esas olarak etkilenen organ sistemleri SSS, göz, kulak ve kalptir:
Klasik semptom üçlüsü (Gregg üçlüsü) konjenital kalp kusurları, iç kulağın sağırlığı ve katarakttan oluşur.
Embriyonun ciddi enfeksiyonları kürtaja yol açabilir. Bununla birlikte, malformasyon olmadan yenidoğan enfeksiyonuna sahip olmak veya doğumdan sonra daha geçici semptomlara sahip olmak da mümkündür.Enfekte yenidoğanların yaklaşık% 10’u hastalığın bir sonucu olarak ölür (mortalite).
Teşhis
Annede karakteristik semptomlar veya anamnestik belirtiler görülmesi durumunda, virüsün kan, idrar veya tükürükten tespit edilmesine çalışılmalıdır. Hamile kadınlarda bağışıklık durumunun hemaglütinasyon inhibisyon testi ve ELISA ile belirlenmesi, yalnızca iki kızamıkçık aşısı yapıldığına dair bir kanıt yoksa endikedir. Yenidoğanda IgM tespiti veya gebeliğin 22. haftasından itibaren bir IgG titresinin belirlenmesi de mümkündür. PCR yoluyla tespit, koryonik villus biyopsisinde veya amniyotik sıvıda gerçekleştirilebilir.
Terapi
Embriyo enfeksiyonundan sonra nedensel tedavi mümkün değildir. Bu nedenle, maternal aşı korumasını sağlayarak kızamıkçık embriyopatisinin önlenmesi çok önemlidir. Bu nedenle, planlanan gebeliklerden önce kızamıkçık titresi belirlenmeli ve koruma yeterli değilse anne yeniden aşılanmalıdır.
Zaten hamile olan bir kadının aşılanması önerilmez. Doğmamış çocuğun aşı virüsü ile enfekte olmasına yol açabilir, ancak genellikle kızamıkçık embriyopatisi ile sonuçlanmaz. Hamile bir kadın kızamıkçık geçiren kişilerle temas ederse, takip eden iki gün içinde kızamıkçık antiserumu ile pasif aşılama yapılmalıdır – ancak etkisi belirsizdir. Annenin IgG antikorları tespit edilirse, aşılama veya önceki hastalığa bağlı bağışıklık varsayılabilir. Hamile kadın 4. ayın sonuna kadar düzenli olarak taze enfeksiyon testi yaptırmalıdır.
Annede kızamıkçık enfeksiyonuna dair kanıt varsa (klinik, seroloji) ve çocukta öngörülebilir malformasyonlar varsa, kürtaj 4. aya kadar düşünülmelidir.
Profilaksi
En etkili profilaksi kızamıkçığa karşı aşılamadır. STIKO’nun güncel aşı önerisine göre kızamık-kabakulak-kızamıkçık aşısı ilk kez 11-14 aylar arasında, ikinci kez ise 15-23 aylar arasında yapılmalıdır.
Hamilelikten önce, hamile kadınlarda kızamıkçık antikor titresi belirlenmelidir. Aşılama yeterli değilse, aşılama hamilelikten önce tekrarlanmalı veya yenilenmelidir. Almanya’da hamile kadınların %7’sinin kızamıkçığa karşı yeterli aşı yaptırmadığı tahmin edilmektedir.
Sinonim: Küme tipi baş ağrısı, cluster headache, Bing-Horton-Syndrom.
Göz ve şakak bölgesinde günde 8 defa (genellikle geceleri) 15-180 dakika süren özellikle şiddetli erkek yan ağrı atakları ile primer baş ağrısı.
Terapi, oksijen inhalasyonu ve triptanlar ile ayrıca verapamil ve prednizolon ile profilaktiktir.
Kaynak: https://66.media.tumblr.com/3a65b32872c45c34ead466c33406ae8a/tumblr_nexkumltjw1qzikspo1_400.jpg
Epidemiyoloji
Küme baş ağrısı, 30 ila 40 yaş arasındaki erkekleri etkileyen nadir bir hastalıktır. Hasta erkeklerin hasta kadınlara oranı 3: 1’dir.
Fizyoloji
Küme baş ağrısı, aynı zamanda paroksismal hemikraniyi ve SUNCT sendromunu da içeren trigemino-otonom baş ağrısı hastalıkları (TAK) (Uluslararası Baş Ağrısı Derneği 2004 Baş Ağrısı Sınıflandırma Komitesi) grubuna aittir. Bunlar ağrı ataklarının süresi, sıklığı, ritmi ve yoğunluğu bakımından farklılık gösterir.
Etiyoloji
Etiyoloji halen belirsizdir (2020). Ağrı ataklarının oluşumu ve sıklığı görünüşte günlük ve mevsimsel etkilerden etkilenir. Bu fenomen ve görüntüleme tekniklerinin (PET) sonuçları nedeniyle, tetikleyici nöronal süreçler hipotalamusun kronobiyolojik kontrol merkezlerinde bulunur.
Alkol, nikotin ve nitrogliser nöbet geçirtebilir. Ek olarak, küme baş ağrısının diğer olası tetikleyicileri de bilinmektedir. Bu tetikleyicilere tepki, kişiden kişiye büyük farklılık gösterir.
Belirtiler
Küme baş ağrısı, bir tarafta frontotemporal olarak lokalizedir ve çok şiddetlidir. Ataklar günde 8 defaya kadar ortaya çıkabilir ve 15 dakika ile 3 saat arasında sürebilir. Semptomlar tipik olarak gece veya sabah erkenden ortaya çıkar. Belirli bir zamanda tekrar eden bir olay da yaygındır.
Etkilenen hastalar ağrı başladığında uyanır ve ağrıyı çok yoğun ve zorlu bir şekilde hisseder. Ağrı genellikle saldırılar sırasında güçlü bir hareket etme dürtüsünü tetikler.
Baş ağrısı aşamaları sırasında, aynı tarafta (‘ipsilateral’) tanı amaçlı karakteristik vejetatif semptomlar görülür:
Konjonktiva artmış vasküler paterni
Artan lakrimasyon
Horner sendromu
Burun mukozasının şişmesi, rinore
Yüzdeki hiperhidroz (alında belirgin)
Göz kapağı ödemi
Hastalıkların çoğu epizodiktir (zamansal ‘kümeler’), yani. nöbetler düzenli olarak (günlük) haftalar ila aylar süresince gerçekleşir, ardından aylar ila yıllar arasında semptomsuz bir aralık gelir. Belirti içermeyen belirgin aralıklar yoksa, kronik bir küme baş ağrısı vardır.
Tanı hastanın tıbbi geçmişine (atağın ifadesi? Provokabilite?) ve mümkünse nöbetin doğrudan gözlemlenmesine dayanır. Nitrogliserin (örneğin dilaltı) kullanılarak bir saldırı tetiklenebilir, ancak provokasyon hastaların en iyi yarısında başarılı olur.
Bir saldırı durumunda, yüksek konsantrasyonlu bir maske ile % 100 oksijen (8-15 l / dak – 15 dakika) uygulanması etkisini kanıtlamıştır.
Şiddetli KOAH bu tedaviye kontrendikedir.
Nazogastrik tüp kullanımı, gerekli akış hızına ulaşılamadığı için küme hastalarında etkisiz olarak tanımlanmıştır. Alternatif olarak, burun spreyi olarak lokal bir anestezik (örn. Lidokain) kullanılabilir.
Sumatriptan veya zolmitriptan da nöbetin geçmesi için kullanılır. Oral ilaçların etkileri çok geç olduğu için bu maddeler deri altına veya nazal olarak uygulanmalıdır.
Verapamil ve / veya kortikoidlerle profilaksi, ataklar veya kronik seyir sırasında endikedir. Bunlar etkili değilse, ikinci seçenek olarak lityum, metisergid veya topiramat kullanılabilir. Diğer önleyici tedavi seçenekleri, melatonin, uzun etkili triptanlar (örn. Naratriptan ve frovatriptan), valproik asit, dihidroergotamin (DHE) IV’tür. Veya pizzotifen. Bir madde tek başına etki etmezse, bir kombinasyon tıbbi gözetim altında test edilmelidir.
SPG stimülasyon terapisi adı verilen yeni birtedavi yöntemi yardım vaat ediyor. Yaklaşık badem büyüklüğünde bir nörostimülatör, sfenopalatin gangliyonun hemen yakınındaki oral mukoza yoluyla oral olarak implante edilir. Küme baş ağrısının gelişimi hala bilinmemekle birlikte, tüm ağrı uyaranlarının bu sinir düğümünden iletildiği bilinmektedir. Hasta, elmacık kemiğinin arkasında tuttuğu bir uzaktan kumanda kullanarak cihazı aktive edebilir. Bu, gangliyona elektriksel uyarılar gönderir ve elektriksel olarak uyarır. Daha sonra bir küme saldırısı takip ederse, sinirler önceki elektriksel uyarılar tarafından aşırı uyarıldığından dolayı ağrı uyaranlarını iletmez.
Araştırmacılar, küme baş ağrılarının hipotalamustan (beynin sirkadiyen saatimizle ilişkili kısmı) kaynaklandığına ve bir hastaya mantar dozu verildiğinde, aktif psilosibinin bölgeye kan akışını yavaşlatarak küme saldırısının başlamasını önlediğine inanıyor. Ancak bariz nedenlerden ötürü, doktorların yapabileceği tek şey bu tedaviyi önermek ve bu arada pek etkisi olmayabilecek hapları yazmaktır.
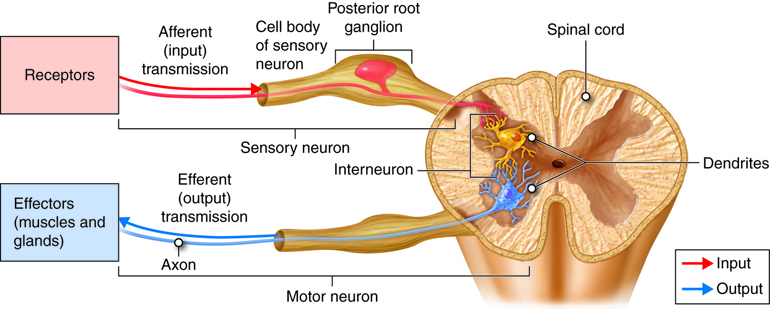
Motor nöronlar, merkezi sinir sistemi (MSS) olarak bilinen beyin ve omurilikten, ön boynuzlarından gelen sinir uyarılarını efektörler olarak bilinen kaslar ve bezler dahil olmak üzere vücudun çeşitli bölgelerine iletmekten sorumlu bir sinir hücresi türüdür.
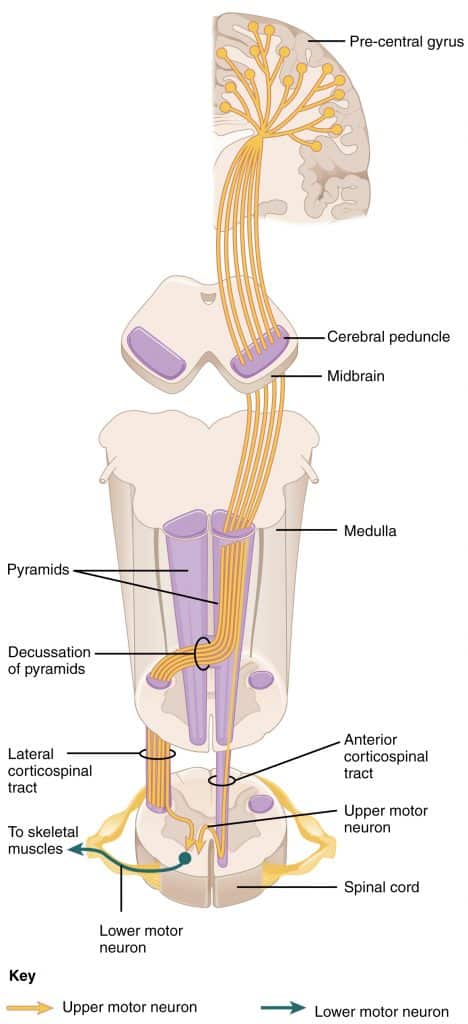
Anatomi ve Yapı
Motor nöronlar iki ana tipe ayrılabilir: üst motor nöronlar (UMN’ler) ve alt motor nöronlar (LMN’ler).
Üst motor nöronlar serebral korteksin motor bölgesinde veya beyin sapında bulunur. Bu bölgelerden gelen sinir uyarılarını omurilikte veya kraniyal sinir çekirdeklerinde bulunan alt motor nöronlara taşırlar.
Alt motor nöronlar ya omurilikte ya da kraniyal sinir çekirdeklerinde bulunur. Üst motor nöronlardan sinyal alırlar ve doğrudan kasları innerve ederek kasılmalarına neden olurlar1.
Her motor nöron bir hücre gövdesi, dendritler ve bir aksondan oluşur.
Hücre gövdesi çekirdek ve diğer organelleri barındırır.
Dendritler, diğer nöronlardan sinyalleri alan ve bunları hücre gövdesine doğru taşıyan kısa, dallanan yapılardır.
Akson, elektriksel uyarıları hücre gövdesinden hedef kas veya beze taşıyan uzun, ince bir çıkıntıdır1.
Motor nöronların aksonları genellikle çok uzundur çünkü MSS’den kontrol ettikleri kas veya beze ulaşmaları gerekir. Bazılarının uzunluğu bir metreye kadar çıkabilir!
Sınıflandırma
Ulaştığı yerlere göre
Somatik Motor nöronlar
İskelet kasına bağlanan motor nöronlardır;
α-Motor nöronu
β-Motor nöronu
γ-Motor nöronu
Viszeral Motor nöronlar
Düz kaslara bağlanan motor nöronlardır;
Özel viseral motor nöronlar (dal motor nöronları)
Genel viseral motor nöronlar
Hiyerarşiye göre
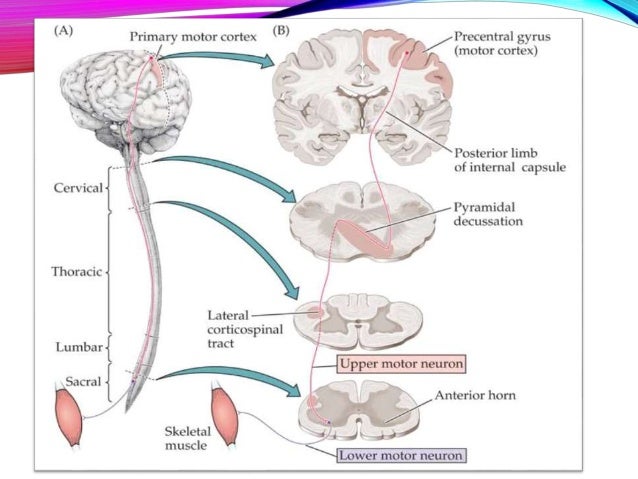
SSS’deki konumlarına göre, somatik motor nöronlar için de bir ayrım yapılır: bir üst motor nöron (1. motor nöron = piramit yörüngesinin 1. nöronu) ve bir alt motor nöronu (2. motor nöron = piramit yolunun 2. nöronu).
Üst motor nöronlar
‘Üst motor nöronlar’ veya UMN’ler olarak da bilinen üst motor nöronları, ağırlıklı olarak serebral korteksin motor merkezi olan motor kortekste bulunur. Bu motor nöronlar gönüllü motor becerilerini kontrol eder. Lifleri kasları doğrudan kontrol etmez, ancak alt motor nöronları aracılığıyla kontrol eder.
Alt motor nöronlar
‘alt motor nöronları’ veya AMN olarak da bilinen alt motor nöronları, iskelet kasını doğrudan innerve eder. Motor çekirdeğini oluşturdukları omuriliğin ön boynuzunda otururlar. Aksonlarınız ön sinir kökleri olarak omurilikten çıkar ve omurilik sinirlerinin bir parçası olarak omuriliği terk eder.
Fonksiyon
Motor nöronlar istemli hareketten sorumludur. Uzantı, serebral korteksin motor bölgelerinde bir motor komutu üretildiğinde başlar. Bu sinyal beyin sapı veya omurilikteki UMN’lerden aşağıya doğru ilerler ve bunlar da LMN’ler üzerinde sinaps yapar. LMN’ler daha sonra sinyali MSS’den efektöre (kas veya bez) gönderir. Sinyal kasa ulaştığında, kasın kasılmasına neden olarak harekete yol açar1.
Motor nöronlar aynı zamanda belirli uyaran türlerine verilen otomatik tepkiler olan reflekslerde de rol oynar.
Hastalıklar ve Bozukluklar
Motor nöronları etkileyebilen bir dizi hastalık ve bozukluk vardır:
Lou Gehrig hastalığı olarak da bilinen Amyotrofik lateral skleroz (ALS), öncelikle motor nöronları etkileyen nörodejeneratif bir hastalıktır. İlerleyici kas güçsüzlüğü ve atrofisine ve nihayetinde felce neden olur2.
Spinal musküler atrofi (SMA), omurilikteki alt motor nöronları etkileyen, kas güçsüzlüğü ve erimesine yol açan genetik bir hastalıktır3.
Motor nöron hastalığı (MND), motor nöronların dejenerasyonu ve kaybı ile karakterize edilen çeşitli durumları (ALS ve SMA dahil) kapsayan bir terimdir4.
Patoloji
Motor nöronlar, birinci, ikinci veya her iki motor nöronu etkileyen çeşitli sinir bozukluklarından etkilenebilir. Bunlar ‘motor nöron hastalıkları‘ (MND) terimi altında özetlenmiştir.
Patoloji
Hastalık
1. Nöron
Kalıtsal Spastik Parapleji (HSP)
2. Nöron
Spinal müsküler atrofisi (SMA)
Her iki Nöron
Amyotrofik Lateral Skleroz (ALS)
Tarih
Motor nöronların tarihi, Fransız nörolog Jean-Martin Charcot’nun amiyotrofik lateral sklerozun (ALS) klinik ve patolojik özelliklerini ilk kez tanımladığı 19. yüzyılın başlarına kadar uzanmaktadır. ALS, istemli kas hareketini kontrol eden hücreler olan motor nöronları etkileyen ilerleyici bir nörodejeneratif hastalıktır.
Charcot’nun çalışmalarını, 1900 yılında “motor nöron hastalığı” terimini ortaya atan William Gowers da dahil olmak üzere diğer nörologların araştırmaları takip etmiştir. 20. yüzyılın başlarında, Alois Alzheimer ve Paul Flechsig gibi nöropatologların çalışmaları, motor nöronların anatomisi ve patolojisini daha iyi anlamamıza yardımcı oldu.
Yirminci yüzyılın ikinci yarısında ALS’nin genetiğini anlamamızda önemli ilerlemeler kaydedilmiştir. 1993 yılında ALS ile bağlantılı ilk gen tanımlandı ve o zamandan bu yana 20’den fazla gen hastalıkla ilişkilendirildi. Bu araştırma, ALS için yeni tanı testlerinin ve tedavi stratejilerinin geliştirilmesine yol açmıştır.
Bugün hala ALS için bir tedavi yoktur, ancak hastalığın ilerlemesini yavaşlatmaya ve ALS’li kişilerin yaşam kalitesini artırmaya yardımcı olabilecek bir dizi tedavi vardır. ALS ile ilgili araştırmalar devam etmektedir ve gelecekte bir tedavi bulunacağına dair umut vardır.
İşte motor nöronların tarihindeki bazı önemli olaylar:
1869: Jean-Martin Charcot ilk kez ALS’nin klinik ve patolojik özelliklerini tanımladı.
1900: William Gowers “motor nöron hastalığı” terimini icat etti.
1919: Alois Alzheimer ve Paul Flechsig motor nöronların anatomisini ve patolojisini tanımlar.
1993: ALS ile bağlantılı ilk gen tanımlandı.
2000s: ALS için yeni tanı testleri ve tedavi stratejileri geliştirildi.
Bugün: ALS için hala bir tedavi yoktur, ancak hastalığın ilerlemesini yavaşlatmaya ve ALS’li kişilerin yaşam kalitesini artırmaya yardımcı olabilecek bir dizi tedavi vardır.
Motor nöronların tarihi uzun ve karmaşıktır, ancak aynı zamanda bir ilerleme öyküsüdür. Bilim insanları ve klinisyenlerin son iki yüzyıldaki çalışmaları sayesinde, artık bu hastalığı ve mevcut potansiyel tedavileri çok daha iyi anlıyoruz.
Kaynakça:
Purves D, Augustine GJ, Fitzpatrick D, et al., editors. Neuroscience. 2nd edition. Sunderland (MA): Sinauer Associates; 2001. The Functional Organization of the Motor System in the Spinal Cord. DOI
Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nature Reviews Neuroscience. 2001 Nov;2(11):806-19. DOI
Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008 Jun 28;371(9630):2120-33. DOI
Talbot K. Motor neuron disease. Postgraduate Medical Journal. 2002 Sep;78(923):513-9. DOI




















Yorum yazabilmek için oturum açmalısınız.